"Huntington's-Disease-Therapeutics"-Konferenz 2018 Tag 3
Neuigkeiten vom dritten Tag der "Huntington's-Disease-Therapeutics"-Konferenz 2018: Huntingtin(-Verminderung)
 Von Dr Jeff Carroll 23. März 2018 Bearbeitet von Professor Ed Wild Übersetzt von Rebecca Ursprünglich veröffentlicht am 2. März 2018
Von Dr Jeff Carroll 23. März 2018 Bearbeitet von Professor Ed Wild Übersetzt von Rebecca Ursprünglich veröffentlicht am 2. März 2018
Guten Morgen vom Abschlusstag der “Huntington’s-Disease-Therapeutics”-Konferenz 2018! Heute gibt es zwei Teilsitzungen, die erste dreht sich um das Eiweiß, das im Auftrag des Huntington-Gens hergestellt wird, die zweite beinhaltet Neues von den Huntingtin-Verminderungsstudien durch Wave Life Sciences und Ionis Pharmaceuticals.
Donnerstagmorgen - das Protein Huntingtin
Jeder Huntington-Patient hat die gleiche Mutation geerbt - eine Erhöhung der C-A-G-Wiederholungen. Diese Verlängerung befindet sich in einer der beiden Kopien des Huntington-Gens und in der ersten Teilsitzung heute geht es um das Huntingtin.

Sandrine Humbert von der Université Grenoble Alpes interessiert sich seit langem für die Entwicklung des Gehirns und wie das Huntington-Gen und Eiweiß diese beeinflussen. Um ein Verständnis zu entwickeln, züchtete Humberts Gruppe eine Maus mit weder gesunden noch mutiertem Huntington-Gen. Sie stellten fest, dass die Gehirnzellen ohne Huntington-Gen sich auf abnormale weise teilten und bewegten. Im frühen Stadium der Hirnentwicklung bewegen sich neu entwickelte Zellen in Richtung ihrem späteren Einsatzort, oftmals entlang von Ketten, die durch andere Zellen gebildet werden. Dieser Prozess wird durch die Löschung des Huntington-Gens verändert, wodurch auf eine wichtige Rolle des Gens bei der Gehirnentwicklung geschlossen wird.
Andrea Caricasole vom IRBM Science Park führt gerade eine großangelegte Studie an kleinen Accessoires durch, mit denen der eigentlichen Bildung des Proteins das Huntingtin dekoriert werden. Mit diesen Accessoires können Zellen die Funktion des Proteins verändern. Abhängig von den vorhandenen Dekorationen reagiert das Huntingtin unterschiedlich auf bestimmte Signale. Es kann sogar sein, dass das mutierte Huntingtin davon abgehalten wird, Zellen zu beschädigen. Darüber haben wir bereits früher berichtet. Caricasoles Team entwickelt äußerst empfindliche Tests für individuelle Accessoires am Huntingtin. So können die Wissenschaftler beobachten, wie und ob sie sich im Verlauf der Krankheit verändern und eventuell Wege finden, sie zu optimieren.
Rohit Pappu von der Washington University verwendet einen gezielten Ansatz, um Huntingtin zu verstehen. In seinem Labor werden Werkzeuge entwickelt, um den Teil des Eiweißes zu verstehen, dessen Form durch das mutierte Huntington-Gen beeinflusst wird. Pappus Team setzt in hohem Maße Computertechnik ein, um eine Vorhersage der Form zu ermöglichen. Bisher wurde eine Art Kaulquappenform beobachtet. Diese Gestalt wurde in der Wissenschaftsgemeinschaft bereits heftig diskutiert.
Xaio-Joang Li von der Emory University hat ein interessantes Mausmodell entwickelt, bei dem das Huntington-Gen in ausgewachsenen Mäusen in Gehirn, Körper oder in beidem ausgeschaltet werden kann. Die Mäuse haben hierbei keine mutierte Kopie des Gens, sondern zwei gesunde. So kann die Funktion des gesunden Gens verstanden werden, beziehungsweise ob dessen Entfernung Konsequenzen hat. Nach der Abschaltung des Gens wurden keine Besonderheiten in den Gehirnen der Mäuse festgestellt, allerdings entzündete sich ihre Bauchspeicheldrüse. Es ist nicht klar, was das genau für Huntington-Patienten bedeutet. Allerdings zielen derzeit laufende Huntingtin-Verminderungsstudien nicht darauf ab, das Huntingtin im Körper zu reduzieren, sondern im Gehirn. Li verwendete auch die Genschere CRISPR-Cas9 um die schädlichen Teile des Huntington-Gens in Mäusen heraus zu schneiden. Die Deaktivierung des mutierten Huntington-Gens in Mäusen führte zur Reduzierung des giftigen Huntingtins und dazu, dass die Mäuse sich besser bewegen konnten. Dr. Li arbeitet also auf verschiedenen Ebenen gleichzeitig. Er hat auch mithilfe der CRISPR-Methode ein Schwein als Huntington-Modell entwickelt. Dieses könnte hilfreich für die Erprobung von Medikamenten sein, da Schweinegehirne den menschlichen recht ähnlich sind.
„Kochanek gelang es, dass Protein einzufrieren. So konnte er per Elektronenstrahl und Computer tausende Bilder davon erstellen. Es handelt sich um die ersten jemals erzeugten Bilder von der Molekularstruktur des Huntingtins. “
Ankur Jain von der University of California in San Diego widmet sich der RNA - dem Botenstoff, der die Instruktionen auf der DNA übernimmt, um Proteine herzustellen. Unsere DNA befindet sich im Zellkern, die RNA allerdings kann sich frei durch die ganze Zelle bewegen. Bisher wurde meist angenommen, dass viele erbliche Nervenkrankheiten durch giftige Eiweiße verursacht werden, mittlerweile gibt es allerdings Hinweise, dass auch Boten-RNAs, die von mutierten Genen erzeugt werden, giftig sein können. Zum Beispiel können sich RNA-Abschnitte an wichtige Enzyme hängen und sie an ihrer Arbeit hindern. Ein Anzeichen für die Giftigkeit der RNA ist die Entstehung von ungewöhnlichen RNA-Tropfen in Zellen von Huntington-Patienten aber auch bei anderen Nervenkrankheiten. Jain hat festgestellt, dass er solche Tropfen auch künstlich aus RNA erzeugen kann, indem er die Substanz erhitzt und abkühlt wie Wackelpudding. Die Tropfen bilden sich nur, wenn die RNA bestimmte Sequenzen enthält, wie die mit den vielen CAG-Wiederholungen bei der Huntington-Krankheit. Noch ist nicht klar, ob sie Schaden anrichten, aber es ist möglich. Zum Beispiel könnte die RNA im Zellkern stecken bleiben und so nicht dazu beitragen, Eiweiße herzustellen. Antisense-Moleküle sind in der Lage sich mit der RNA zu verbinden und die Tropfenbildung zu verhindern. Auch andere Medikamente könnten in Frage kommen, um einer Verklebung von RNA entgegenzuwirken.
Ein aufregender Spezialbeitrag folgt von Stefan Kochanek, dessen Forscherteam gerade die Struktur des Huntingtin-Proteins entschlüsselt hat! Herauszufinden, wie Eiweiße aussehen ist ein wichtiger Schritt, um zu verstehen wie sie funktionieren und wie ihre Funktion durch Medikamente verändert werden könnte. Das Huntington-Gen als Verursacher der Huntington-Krankheit wurde vor 25 Jahren isoliert, das erzeugte Protein ist allerdings groß und sehr komplex, was die Entschlüsselung der Struktur sehr lange herausgezögert hat. In der Vergangenheit haben Forscher das Eiweiß sogar in den Weltraum geschickt, um zu testen, ob es dort eine kristalline Struktur entwickelt, aber ohne Erfolg. Kochaneks Gruppe konnte also erreichen, was andere nicht schafften und ihre Ergebnisse wurden vor kurzem in der Wissenschaftszeitschrift “Nature” veröffentlicht. Der große Schlag gelang, als ein weiteres Protein zur Stabilisierung von Huntingtin identifiziert wurde, sein Name ist HAP40 (“Huntingtin-assoziiertes Protein 40”). Nach der Stabilisierung mit HAP40 fror Kochanek das Protein ein und nutzte einen Elektronenstrahl, um die ersten verfügbaren Bilder der Molekularstruktur zu erzeugen. Das ist richtig klasse und liefert eine Menge Information, mit der weitergearbeitet werden kann. Einen Vorbehalt gibt es allerdings: einige Regionen des Proteins waren auch nach der Stabilisierung noch zu weich, um ihre Struktur zu untersuchen - unter diesen Regionen ist auch die wirklich wichtigste, nämlich die mit der Mutation.
Donnerstagnachmittag - Huntingtin-Verminderung
Wir kommen zum herausragenden Ende des Tages und der Konferenz mit dem Abschnitt zu den Huntingtin-Verminderungs-Studien. Bei diesem Therapieansatz wird versucht, die Menge des produzierten Huntingtins zu reduzieren. Hierfür gibt es unterschiedliche Herangehensweisen, oft wird mit der Boten-RNA gearbeitet, die die Informationen des Huntington-Gens zum Bau des Eiweißes überbringt.
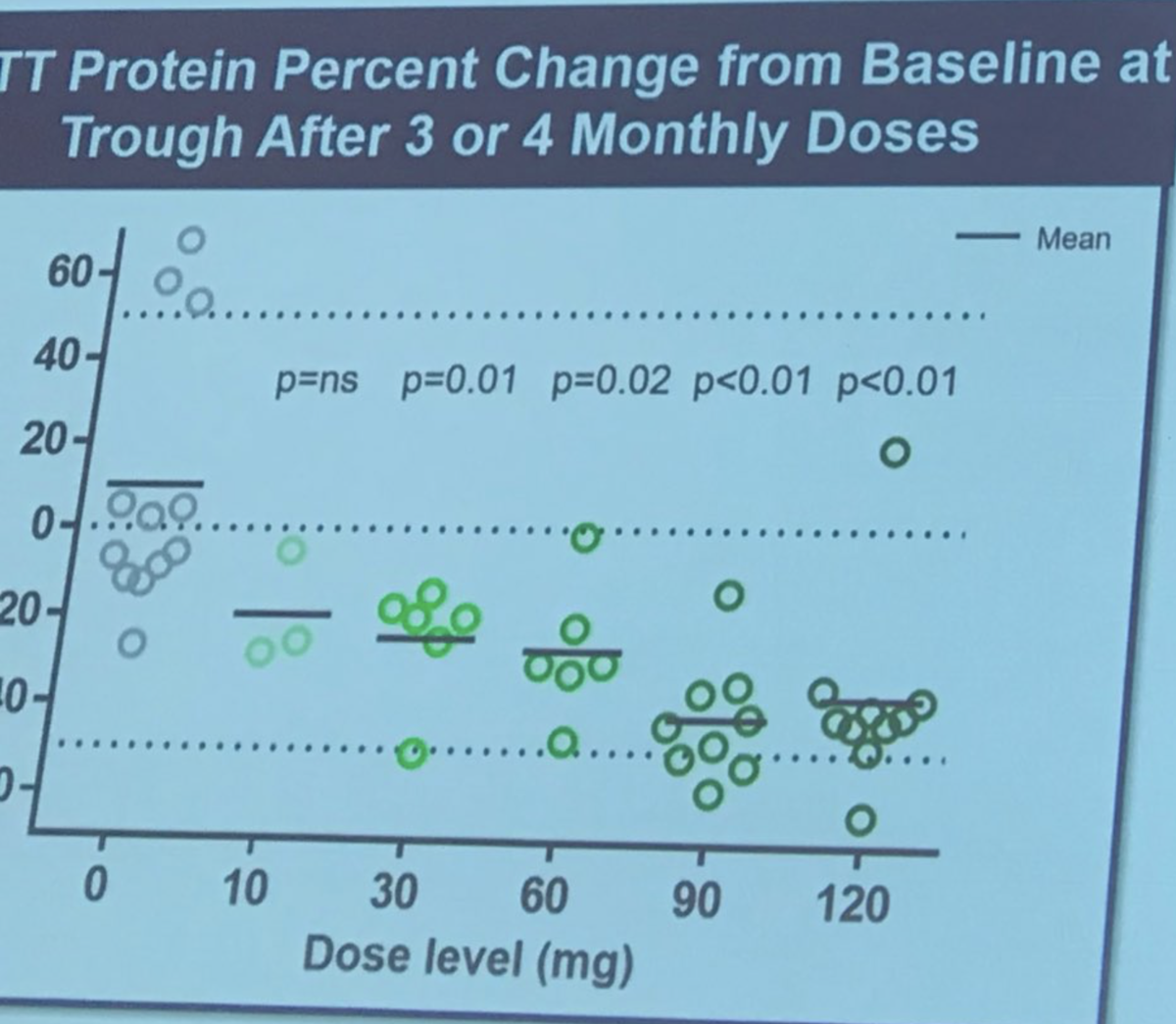
Michael Rape von der University of California in Berkeley versucht Zellen dazu zu bringen, bestimmte Proteine in ihrem Innern zu zerstören. In vielen Fällen, auch bei der Huntington-Krankheit, wäre das selektive Entfernen spezifischer Eiweiße sehr hilfreich. Es gibt für Zellen mehrere Wege, um Proteine abzubauen - einer der wichtigsten verwendet dazu eine winzige chemische Kennzeichnung, die als “Ubiquitin” bezeichnet wird. Diese erkennen Zellen als eine Art “iss-mich”-Signal und bauen die betroffenen Eiweiße ab. Rapes Gruppe war daran beiteiligt, zu verstehen, wie Zellen diese Ubiquitin-Markierungen verwenden, um Eiweiße zu kennzeichnen, die sehr schnell vernichtet werden müssen - zum Beispiel solche, die giftig für den Körper sind. Sie haben Werkzeuge entwickelt, mit denen Wissenschaftler erstmals dabei zusehen können, wie Proteine diesen zügigen Prozess durchlaufen. Die Maschinerie für den Eiweißabbau ist machtvoll und Rapes Gruppe möchte sie nutzen. Eine kürzlich entwickelte Technik namens “PROTAC” erlaubt es Forschern, das Ubiquitin gezielt zur Zerstörung spezieller Proteine einzusetzen.
Scott Zeitlin von der University of Virginia arbeitet mit Huntington-Mäusen, um herauszufinden, wozu es führt, wenn man das mutierte Huntingtin, das gesunde Huntingtin oder beide minimiert. Seien Sie daran erinnert, dass jeder Mensch eine Kopie des Huntington-Gens von jedem Elternteil erbt - und dass die meisen Menschen mit der Huntington-Krankheit eine gesunde und eine mutierte Kopie besitzen. Wissenschaftler bezeichnen das gesunden Huntingtin als den “wilden” Typ, da es in der Wildnis häufiger vorkommt. Diese Tatsachen sind grundlegend, denn alle Huntingtin-Verminderungsstudien zielen darauf ab, die Gesamtmenge von Huntingtin im Gehirn zu reduzieren. Einige, wie das Medikament von Ionis, verringern beide Versionen des Proteins gleichermaßen. Andere, wie Wave’s Medikamente, setzen den Gehalt des mutierten Proteins stärker herab als den des wilden Typs. Wir gehen davon aus, dass die eine oder die andere Möglichkeit heilsam wirken wird - allerdings ist die Frage nach der Sicherheit der Methoden immer noch offen. Zeitlin hat Mäuse gezüchtet, bei denen die Produktion des mutierten, des wilden oder beider Proteine verringert werden kann, wenn sie ausgewachsen sind. Zeitlin fand heraus, dass eine frühe Verringerung des mutierten Huntingtins einen größeren Effekt auf die Ansammlung desselben im Gehirn hatte. Auf ähnliche Weise, wurden auch Gewichtsverlust und Bewegungsfähigkeiten bei früherer Reduzierung stärker beeinflusst. Das gleiche geschah, wenn er beide Proteine verringerte - wenn die Behandlung früh startete, waren die positiven Auswirkungen größer. Zusammengefast, je früher desto besser, wenn von der Unterdrückung der Huntingtinproduktion die Rede ist. Bei einem Test (Test der Greifstärke), verbesserte die Verringerung des mutierten Proteins alleine die Leistungsfähigkeit, wohingegen die Verrringerung beider Versionen dies nicht tat. Bei den anderen Tests zeigten sich beide Varianten in etwa gleich wirkungsvoll mit dem Schlüsselfaktor des Zeitpunkts des Behandlungsbeginns. Zeitlin beobachtete auch, was passiert, wenn man die Bildung von Huntingtin erneut zulies, und das stellte sich aus schädlich für die Mäuse heraus. Daraus lässt sich schließen, dass eine Langzeitanwendung besser ist als über einen kurzen Zeitraum.
Jodi McBride vom OHSU Gesundheitszentrum in Oregon stellt ihre Arbeit vor: sie verwendet harmlose Viren, um Anleitungen in Gehirnzellen zu transportieren, die ihnen helfen ihre eigenen RNA-zerstörenden Moleküle herzustellen. Eins der Vorteile dieses Ansatzes ist, dass die RNA-zerstörenden Moleküle dann für immer hergestellt werden können, sodass eventuell nur eine einmalige Behandlung notwendig ist. McBride testet die Behandlung gerade an Affen, sie haben große und komplexe Gehirne, die dem meschlichen ähnlich sind. Im Speziellen arbeitet ihr Team daran, das Virus in ein Hirnareal namens “Putamen” zu befördern. Es handelt sich dabei um eine der verwundbarsten Hirnregionen bei der Huntington-Krankheit - das Putamen ist ausgelöst durch die Genmutation am stärksten von der Schrumpfung betroffen. McBride beschreibt Fortschritte bei der Operation, die für die Viruseinpflanzung nötig ist. Sie verwendet bildgebende Methoden wie MRI, um zu sehen, was passiert, wenn die Injektion stattfindet. Die Virusbehandlung führte zu Reduzierungen der RNA des Huntington-Gens um etwa die Hälfte im gesamten Putamen, eine merkliche Verbesserung gegenüber früheren Versuchen.
Mike Panzara von der Firma Wave Life Sciences spricht über zwei Studien mithilfe von “Antisense Oligonukleotiden” (ASOs). ASOs sind kurze, veränderte Stücke von DNA, die in Zellen vordringen und eine bestimmte RNA gezielt vernichten, sodass sich die Menge des produzierten Eiweißes verringert. Warum führt Wave zwei Studien durch? Wave’s Ansatz beruht auf der gezielten Ansteuerung winziger genetischer Variationen namens SNPs (sprich: “Snips”) im Huntington-Gen. Durch diese Variationen wird die Huntington-Krankheit nicht ausgelöst, sie sind einfach Teil der normalen genetischen Vielfalt unter uns Menschen. Interessanterweise befinden sich die gleichen SNPs immer nur auf einer der beiden Kopien des Huntington-Gens, die jeder Mensch hat. Indem die Variationen angesteuert werden, kann Wave also festlegen, welche der beiden Genkopien anvisiert wird: die mutierte oder die gesunde. Momentan führt Wave frühe Studien zur Unbedenklichkeit zweier ASOs durch, die Studien heißen PRECISION-HD1 und PRECESION-HD2. Ziel der ASOs ist jeweils eine andere Variation im Huntington-Gen. Leicht verzwickt an diesem Ansatz ist also, dass die Patienten als Voraussetzung nicht nur das mutierte Huntington-Gen sondern auch bestimmte SNPs als Begleiterscheinung mitbringen müssen, damit diese gezielt angesteuert werden können. Wave hat neue Technologien entwickelt, die in der Lage sind, diese Variationen zu erkennen und festzustellen, welche sich auf der mutierten Kopie des Gens befinden. In einer Vorstudie konnte Wave bei 64% der Freiwilligen entweder die in PRECISION-HD1 oder die in PRECESION-HD2 gesuchte Variation identifizieren.
„Spontaner Beifall als Tabrizi den mutigen Freiwilligen der ersten Studie dankt und sie als “wahre Helden der Wissenschaft” bezeichnet. “
Anne Smith von der Firma Ionis und Sarah Tabrizi vom University College London präsentieren die Ergebnisse einer Studie, die ASOs testen sollte, auf die beide Kopien des Huntington-Gens ansprechen. Hier werden nun die Früchte langjähriger Arbeit geerntet: Smith erinnert die Zuhörer, das das Programm von Ionis bereits im Jahr 2005 begann! Sie fingen mit Zell- und Tierstudien an, die frühe Hinweise auf die Reduktion des Huntingtins durch die ASO-Behandlungen lieferten und die Symptome der Huntington-Krankheit abmilderten. In den Jahren 2012 und 2013 wurden Ergebnisse aus Mausmodellstudien veröffentlicht, die zeigten, dass auch hier die Huntingtin-Verminderung die Symptome verbesserte. Smith gibt eine Abriss der Denkweise von Ionis Pharma, die zu den Entscheidung führte, beide Kopien des Huntington-Gens anzuvisieren und nicht bloß die mutierte. Ein Vorteil von ASOs ist, dass sie sich gleichmäßig im ganzen Gehirn verteilen. Smith zeigt Daten aus Experimenten mit Affen, die verdeutlichen, dass sich nach der Injektion in die Gehirn-Rückenmark-Flüssigkeit die ASOs im gesamten Gehirn ausbreiten. Ionis untersuchte dies auch bei größeren Tieren, wie Schweinen mit dem gleichen Ergebnis. Danach wurden Studien zur Giftigkeit bzw. Verträglichkeit durchgeführt, die ergaben, dass die Langzeitanwendung des Medikamentes gut vertragen wurde: bei den Affen ging die Studie über 15 Monate. Es ist nicht möglich, Proben vom Gehirngewebe von Patienten zu nehmen, also wie kann man herausfinden, ob das Medikament wirkt? Smith stellt Ergebnisse aus den Affenstudien vor, bei denen eine Beziehung zwischen der Verringerung des Huntingtins im Gehirn und in der Hirn-Rückenmarks-Flüssigkeit festgestellt wurde. Auf Grundlage dieser Daten hat Ionis ein Computerprogramm entwickelt, das die Vorhersage der Huntingtinverminderung im Gehirn aufgrund einer Lumbalpunktion ermöglicht. Ab diesem Zeitpunkt begann der große Pharmakonzern Roche mit Ionis zusammenzuarbeiten. Roche hat die Ressourcen und die Erfahrung, um mit ASOs komplexe Studien am Menschen durchzuführen.
Sarah Tabrizi vom University College London übernimmt danach, um die erste am Menschen durchgeführte klinische Studie von Ionis/Roche’s ASO-Behandlung vorzustellen. Hierbei handelt es sich um eine Verträglichkeits- und Unbedenklichkeitsstudie. Die Studie wurde an neun Orten im Groß-Britannien, Deutschland und Kanada durchgeführt. Die ASOs wurden den Patienten über Lumbalpunktion injiziert, während die Dosis bei den früheren Teilnehmern geringer und bei den späteren Teilnehmern immer höher angesetzt wurde. Diese vorsichtige Steigerung der Dosis wird vorgenommen, um eine Sicherheitsbewertung der Medikation durch weitere Ärzte zu ermöglichen, die von der Studie unabhängig sind. An der Studie nahmen 46 Freiwillige teil, die sich dem Risiko stellten, als erste Menschen, das Medikament zu erhalten. Die Wissenschaftler konnten die Gehalte von Huntingtin in der Gehirn-Rückenmark-Flüssigkeit messen, da sie, wie vorher beschrieben, die Korrelation mit den Werten im Gehirn nachgewiesen hatten. Aufgrund des von Ionis entwickelten Models, kann davon ausgegangen werden, dass die Huntingtin-Verminderung im Gehirn recht hoch ist. Die Stärke der Reduktion in der Gehirn-Rückenmark-Flüssigkeit ist jedenfalls beachtlich - durchschnittlich minus 40 bis 50 %! Tabrizi erwähnt, dass die Forscher davon ausgehen, dass sich die Menge über eine Anwendungsdauer von sechs Monaten hinweg weiter reduzieren könnte. Tabrizi sagt abschließend: “Das Medikament stellte sich in allen angewendeten Dosen als sicher und gut verträglich heraus”. Ein toller Erfolg! Alle Teilnehmer befinden sich jetzt in einer Verlängerungsstudie, die ohne Placebo-Kontrolle durchgeführt werden kann, sodass sich die Patienten sicher sein können, dass sie tatsächlich das Medikament bekommen. Sie werden weiterhin von den Wissenschaftlern untersucht. Spontanen Beifall gibt es, als Tabrizi den mutigen Freiwilligen der ersten Studie dankt und sie als “wahre Helden der Wissenschaft” bezeichnet.
Was für eine tolle Art das Treffen zu beenden - unglaublich aufregende Zeiten stehen uns bevor, während Roche und Ionis bereits die nächste Studie planen, die dafür ausgelegt sein wird, zu erproben, ob ihr Medikament die Huntington-Symptome bei einer größeren Anzahl von Patienten verbessert.
Update: [Mitteilung an die Huntington-Gemeinschaft] von Ionis (http://hdsa.org/wp-content/uploads/2018/03/Ionis-Roche-CHDI-HD-Community-Statement_01Mar2018.pdf) on the results.


