
Therapiekonferenz zur Huntington-Krankheit 2018 – Tag 3
Aktuelles von Tag 3 der Therapiekonferenz zur Huntington-Krankheit: Huntingtin-Protein – und dessen Senkung

Guten Morgen vom letzten Tag der HD-Therapiekonferenz 2018! Heute gibt es zwei Sitzungen, die erste konzentriert sich auf das Protein, das aus dem HD-Gen hergestellt wird. Die zweite beinhaltet Neuigkeiten zu Huntingtin-senkenden Studien von Wave Life Sciences und Ionis Pharmaceuticals.
Donnerstagmorgen – Huntingtin-Protein
Jeder HD-Patient hat die gleiche Mutation geerbt – eine Verlängerung der Sequenz C-A-G. Diese Expansion findet in einem Gen statt, das wir jetzt HD-Gen nennen. Gene werden von Zellen als Anweisungen zur Herstellung von Proteinen verwendet – die erste Sitzung heute konzentriert sich auf das HD-Protein.

Sandrine Humbert, Universität Grenoble Alpes, hat langjähriges Interesse an der Entwicklung des Gehirns und daran, wie das HD-Gen und -Protein diesen Prozess beeinflussen. Um diesen Prozess zu verstehen, entwickelte Humberts Labor eine Maus, der das HD-Gen und -Protein in ihren Gehirnen fehlte. Sie entdeckten, dass sich Zellen ohne das HD-Gen abnormal teilten und bewegten. Während der Gehirnentwicklung kriechen neu geborene Zellen zu ihrem richtigen Ort und klettern oft an „Seilen“ entlang, die von anderen Zellen gebildet werden. Dieser Prozess wird verändert, wenn das HD-Gen gelöscht wird, was auf wichtige Rollen des HD-Gens in diesem Prozess hindeutet.
Andrea Caricasole, IRBM Science Park, führt eine groß angelegte Studie über „posttranslationale Modifikationen“ des Huntingtin-Proteins durch. Dies bezieht sich auf winzige chemische „Verzierungen“ des Huntingtin-Proteins. Diese Verzierungen ermöglichen es Zellen, die Funktion von Proteinen anzupassen. Das Huntingtin-Protein zum Beispiel hat wahrscheinlich Dutzende dieser Markierungen, die hinzugefügt und entfernt werden und die Funktion von Huntingtin als Reaktion auf eine Vielzahl von Signalen anpassen. Viele dieser Verzierungen bewirken faszinierende Dinge mit dem Huntingtin-Protein und können sogar verhindern, dass mutiertes Huntingtin-Protein Zellen schädigt. Wir haben darüber bereits auf HDBuzz berichtet. Caricasoles Team entwickelt sehr empfindliche Tests für individuelle Huntingtin-Protein-Verzierungen. Diese ermöglichen es ihnen, zu verfolgen, welche davon im Verlauf der Krankheit verändert werden, und vielleicht nach Wegen zu suchen, sie zu beheben.
Rohit Pappu, Washington University, verfolgt einen sehr fokussierten Ansatz zum Verständnis des Huntingtin-Proteins. Sein Labor entwickelt Werkzeuge, um den Teil des Proteins zu untersuchen, dessen Form durch die HD-Mutation beeinflusst wird. Pappus Labor nutzt massive Rechenleistung, um die Form des durch die Mutation veränderten Teils des Huntingtin-Proteins vorherzusagen. Diese Techniken ermöglichen es ihnen, eine „Kaulquappen“-Form zu beobachten. Die Form dieser Kaulquappe war Gegenstand intensiver Debatten im HD-Feld! Pappus Techniken unterstützen eine Seite dieser Debatte nachdrücklich, was uns sicherlich helfen wird, diesen kritischen Teil des Huntingtin-Proteins besser zu verstehen.
Xaio-Joang Li von der Emory University hat ein interessantes Mausmodell entwickelt, bei dem das Huntingtin-Gen bei ausgewachsenen Mäusen im Gehirn, Körper oder beidem ausgeschaltet werden kann. Diese Mäuse haben keine expandierten Huntingtin-Gene – sie helfen uns lediglich zu verstehen, ob das Ausschalten der „gesunden“ Version des Gens Konsequenzen hat. Beruhigenderweise passiert nichts Schlimmes im Gehirn, wenn das Gen ausgeschaltet wird. Unerwarteterweise führte das Ausschalten des Gens zu einer Entzündung der Bauchspeicheldrüse. Es ist unklar, was das für Patienten bedeuten könnte, aber aktuelle Huntingtin-senkende Behandlungen sollen die Huntingtin-Spiegel im Körper nicht signifikant reduzieren – nur im Gehirn. Li hat auch die CRISPR-Cas9-Genom-Editierung eingesetzt, um das schädliche Stück des HD-Gens in Mäusen herauszuschneiden. Die Deaktivierung des mutierten Gens in Mäusen reduzierte erfolgreich die Bildung des toxischen Huntingtin-Proteins, und die Mäuse bewegten sich auch besser. Dr. Li war sehr beschäftigt! Er hat auch ein Huntington-Krankheitsmodell-Schwein mittels CRISPR-Genom-Editierung erstellt. Dies könnte nützlich sein für die Testung neuer Medikamente, da das Schweinegehirn dem menschlichen ähnelt.
„Kochanek fror das Protein ein und nutzte einen Elektronenstrahl, um Tausende von Fotos davon zu machen. Diese wurden dann per Computer kombiniert, um die ersten Bilder der detaillierten molekularen Struktur des Huntingtin-Proteins zu erstellen.“
Ankur Jain von @UCSanDiego untersucht RNA – die „Botenmoleküle“, die entstehen, wenn eine Zelle die Anweisungen in der DNA nutzen möchte, um ein Protein herzustellen. Unsere DNA lebt im Zellkern unserer Zellen, aber RNA schwimmt frei in der gesamten Zelle herum. Die traditionelle Denkweise über viele genetische Hirnerkrankungen ist, dass sie durch toxische Proteine verursacht werden, aber es gibt zunehmend Hinweise darauf, dass manchmal auch die RNA-Botenmoleküle, die aus mutierten Genen produziert werden, toxisch sein können. Zum Beispiel können einige RNA-Sequenzen an wichtige Proteinmaschinen haften und sie daran hindern, ihre Aufgaben in der Zelle zu erfüllen. Ein mögliches Zeichen für toxische RNA ist die Bildung abnormaler RNA-Klumpen, die in Zellen bei HD und anderen Hirnerkrankungen beobachtet werden. Jain hat entdeckt, dass er künstliche RNA-Klumpen bilden kann, indem er sie wie Wackelpudding erhitzt und abkühlt. Diese Klumpen bilden sich nur, wenn die RNA klebrige Sequenzen enthält, wie die aus dem CAG-Bereich bei HD. Es ist unklar, ob diese RNA-Klumpen bei HD Schaden anrichten, aber es ist möglich. Wenn die RNA zum Beispiel im Zellkern festsitzt, kann sie nicht zur Proteinherstellung verwendet werden. Antisense-Moleküle (ähnlich denen, die derzeit in HD-Humanstudien getestet werden) können an die RNA im Zellkern haften und deren Klumpenbildung verhindern. Andere Medikamente könnten theoretisch auch eingesetzt werden, um das Problem der RNA-Klebrigkeit bei Hirnerkrankungen anzugehen.
Spannender, aktueller Vortrag jetzt von Stefan Kochanek, dessen Labor gerade die Struktur des Huntingtin-Proteins entschlüsselt hat! Herauszufinden, wie Proteine aussehen, ist ein wirklich wichtiger Schritt, um zu verstehen, wie sie funktionieren und wie man das mit Medikamenten ändern kann. Das Huntingtin-Gen wurde vor 25 Jahren entdeckt, aber das Protein ist groß, wackelig und klebrig, was es sehr schwierig gemacht hat, seine Struktur zu entdecken. Ein Team schickte das Protein sogar ins All, um zu versuchen, Kristalle zu bilden, aber leider ohne Erfolg. Kochaneks Team war erfolgreich, wo andere gescheitert sind, und ihre Ergebnisse wurden gerade in Nature veröffentlicht. Der große Durchbruch war die Stabilisierung von Huntingtin mit einem anderen Protein namens HAP40 („Huntingtin-assoziiertes Protein 40“). Einmal mit HAP40 stabilisiert, fror Kochanek das Protein ein und nutzte einen Elektronenstrahl, um Tausende von Fotos davon zu machen. Diese wurden dann per Computer kombiniert, um die ersten Bilder der detaillierten molekularen Struktur des Huntingtin-Proteins zu erstellen. Das ist wirklich cool und gibt uns eine Menge zu tun. Ein Vorbehalt jedoch: Einige Bereiche waren immer noch zu wackelig, um die Struktur zu entschlüsseln – einschließlich des überaus wichtigen Teils am Anfang des Proteins, der die Mutation enthält.
Donnerstagnachmittag – Huntingtin-Senkung
Großer Abschluss des Tages und der Konferenz, während die Sitzung zu Huntingtin-senkenden Therapien beginnt. Huntingtin-Senkung bezieht sich auf Ansätze, die darauf abzielen, die Spiegel des Huntingtin-Proteins zu senken. Es gibt viele Wege, dies zu tun, aber viele davon zielen auf die „RNA“ ab, die ein Zwischenprodukt zwischen der Information im HD-Gen und dem Huntingtin-Protein ist.
Michael Rape, UC Berkeley, ist daran interessiert, Zellen dazu zu bringen, einzelne Proteine in einer Zelle zu zerstören. In vielen Fällen, einschließlich HD, wäre es wirklich hilfreich, ein spezifisches Protein selektiv zu entfernen. Zellen haben mehr als einen Proteinabbauweg – ein wichtiger verwendet eine winzige chemische „Verzierung“ namens „Ubiquitin“ als Markierung. Zellen erkennen Ubiquitin als eine Art „Friss mich“-Signal und bauen Proteine ab, die es tragen. Rapes Labor war daran beteiligt zu verstehen, wie Zellen Ubiquitin-Tags verwenden, um Proteine zu markieren, die sehr schnell abgebaut werden müssen – eines, das zum Beispiel toxisch sein könnte. Rapes Labor hat Werkzeuge entwickelt, die es Forschern zum ersten Mal ermöglichen, Proteine bei diesem schnellen Abbauweg zu beobachten. Die Maschinerie für den schnellen Proteinabbau ist ein mächtiges Werkzeug – eines, das Rapes Labor nutzen möchte. Eine kürzlich entwickelte Technik – genannt „PROTAC“ – ermöglicht es Forschern, das Ubiquitin-System zu nutzen, um Zellen dazu zu bringen, spezifische Proteine zu zerstören.
Scott Zeitlin (University of Virginia) arbeitet mit HD-Mäusen, um herauszufinden, was passiert, wenn wir mutiertes Huntingtin, normales Huntingtin oder beides senken. Man muss bedenken, dass jede Person ein Huntingtin von jedem Elternteil erbt – und die meisten Menschen mit HD haben eine normale und eine mutierte Kopie. Wissenschaftler nennen das gesunde/normale Protein „Wildtyp“, weil es das ist, das in der Natur häufiger vorkommt. Diese Fragen sind wichtig, weil alle Huntingtin-senkenden Therapien darauf abzielen, die Gesamtmenge des Huntingtin-Proteins im Gehirn zu reduzieren. Einige, wie das Medikament von Ionis, reduzieren beide Versionen des Proteins gleichermaßen. Andere, wie die Medikamente von Wave, versuchen, das mutierte Protein stärker zu senken als das Wildtyp-Protein. Wir halten es für wahrscheinlich, dass die Senkung des mutierten Proteins allein oder parallel zum Wildtyp-Protein vorteilhaft sein wird – aber es ist immer noch eine offene Frage, ob die Senkung von Huntingtin sicher ist. Zeitlin hat Mäuse gezüchtet, bei denen die Produktion des mutierten, Wildtyp- oder beider Proteine reduziert werden kann, nachdem die Maus ausgewachsen ist. Zeitlin fand heraus, dass eine frühe Senkung des mutierten Huntingtins einen größeren Effekt auf die Anreicherung des Proteins im Gehirn hatte. Ähnlich hatte eine frühe Reduktion des mutierten Huntingtins größere Vorteile bei Gewichtsverlust und Bewegungsfähigkeit der Mäuse. Dasselbe galt für die Reduzierung der Produktion beider Versionen des Proteins – eine frühe Behandlung hatte größere Vorteile. Fazit: Früher ist besser, wenn es darum geht, Huntingtin zu unterdrücken. Bei einem Test (Greifkraft) verbesserte die Senkung nur des mutierten Proteins die Leistung, aber die Unterdrückung beider Versionen nicht. Ansonsten waren beide Ansätze ungefähr gleich wirksam, und der Schlüsselfaktor war, wie früh die Behandlung erfolgte. Zeitlin untersuchte auch, was passiert, wenn man Huntingtin sich erholen lässt, und das war schlecht für die Mäuse. Das deutet darauf hin, dass eine Langzeitbehandlung besser ist als eine Kurzzeitbehandlung – genau das, was man erwarten würde.
Jodi McBride, OHSU, beschreibt ihre Arbeit, bei der sie harmlose Viren einsetzt, um Anweisungen an Gehirnzellen zu liefern, die ihnen helfen, ihre eigenen RNA-zerstörenden Moleküle herzustellen. Einer der Vorteile dieses Ansatzes ist, dass die Viren es ermöglichen, die RNA-zerstörenden Moleküle theoretisch für immer herzustellen, was eine einmalige Behandlung ermöglicht. McBride untersucht ihre Behandlung, indem sie sie an Affen verabreicht, die große, komplexe Gehirne haben, die unseren viel ähnlicher sind. Speziell arbeitet ihr Team daran, das Virus an einen Teil des Gehirns zu liefern, der „Putamen“ genannt wird. Das Putamen ist besonders interessant, da es eine der anfälligsten Hirnregionen bei HD ist – es leidet unter einer starken Schrumpfung bei Menschen, die die HD-Mutation erben. McBride beschreibt Verbesserungen bei der für die Virusverabreichung erforderlichen Gehirnoperation, einschließlich der Verwendung von MRT zur Bildgebung des Gehirns während der Injektionen. Die virale Behandlung führte zu einer Reduzierung der HD-Gen-RNA um etwa die Hälfte im gesamten Putamen, eine bemerkenswerte Verbesserung gegenüber früheren Versuchen. Als Nächstes – Mike Panzara von Wave Life Sciences, die 2 Studien mit „Antisense-Oligonukleotiden“ (ASOs) für HD planen. ASOs sind kurze, modifizierte DNA-Stücke, die in Zellen eindringen und eine Ziel-RNA zerstören, wodurch die Spiegel des Zielproteins reduziert werden.
Panzara erzählt dem Publikum, dass Wave derzeit zwei Studien mit ASOs bei HD-Patienten durchführt. Warum zwei? Waves Ansatz basiert auf der gezielten Bekämpfung winziger genetischer Variationen – genannt SNPs oder „Snips“ – im HD-Gen. Diese winzigen Variationen verursachen keine HD, sie sind nur Teil der normalen genetischen Variation zwischen Menschen – der Grund, warum wir nicht alle eineiige Zwillinge sind. Interessanterweise finden sich diese Varianten nur auf einer der 2 Kopien des HD-Gens, die jede Person besitzt. Durch die gezielte Bekämpfung dieser Varianten können Waves ASOs zwischen den mutierten und nicht-mutierten Kopien des HD-Gens unterscheiden. Wave führt derzeit frühe Sicherheitsstudien mit 2 ASOs in Studien namens PRECISION-HD1 und PRECISION-HD2 durch. Die in diesen Studien verwendeten ASOs zielen auf unterschiedliche genetische Variationen im HD-Gen ab. Der Trick bei diesem Ansatz ist, dass Menschen nicht nur die HD-Mutation geerbt haben müssen, sondern auch begleitende Varianten, die es ermöglichen, die mutierte Kopie des Gens einzigartig zu targetieren. Diese Studien konzentrieren sich also notwendigerweise auf Patienten, die diese Variationen tragen. Wave hat wirklich coole neue Technologien entwickelt, um diese Varianten zu erkennen und zu bestimmen, welche sich auf der mutierten Kopie des HD-Gens befinden, nicht auf der normalen Kopie. Wave führte eine Vorstudie durch, in der sie in 64 % der Freiwilligen Ziele für ihre ASOs finden konnten.
„Spontaner Applaus, als Tabrizi den mutigen Freiwilligen der ersten Studie dankt und sie als „wahre Forschungshelden“ bezeichnet.“
Als Nächstes präsentieren Anne Smith von Ionis und Sarah Tabrizi vom UCL die Ergebnisse einer Studie, die darauf abzielt, ASOs zu testen, die beide Kopien des HD-Gens ansteuern. Dies ist der Höhepunkt jahrelanger Arbeit – Smith erinnert das Publikum daran, dass das Ionis-Programm 2005 begann! Sie begannen mit Zell- und Tierstudien, die frühe Hinweise darauf lieferten, dass ASO-Behandlungen das Huntingtin-Protein reduzieren und HD-ähnliche Symptome verbessern. In den Jahren 2012 und 2013 wurden Ergebnisse von HD-Mausmodellstudien veröffentlicht, die zeigten, dass die Huntingtin-Senkung HD-ähnliche Symptome verbesserte. Smith erläutert die Logik, die @ionispharma verwendete, um die Entscheidung zu treffen, ASOs einzusetzen, die beide Kopien des HD-Gens ansteuern, anstatt nur die mutierte Kopie. Ein Vorteil von ASOs ist, dass sie sich weit im gesamten Gehirn verteilen. Smith zeigt Daten aus Affenexperimenten, die belegen, dass sich ASOs nach Injektion in die Rückenmarksflüssigkeit sehr weit im gesamten Gehirn verteilen. Ionis untersuchte auch die Verteilung bei noch größeren Tieren, wie Schweinen, und stellte fest, dass das Medikament sehr weit verteilt war. Anschließend wurden Toxizitätsstudien durchgeführt, die darauf hindeuteten, dass die Langzeitverabreichung des Medikaments sehr gut vertragen wurde (bis zu 15 Monate in Affenstudien). Es ist fast unmöglich, Hirngewebe von Patienten zu entnehmen, die mit ASOs behandelt wurden – wie werden wir also wissen, ob das ASO seine Aufgabe erfüllt hat? Smith beschreibt Affenstudien, die eine Beziehung zwischen der Huntingtin-Senkung im Gehirn und der Senkung in der Rückenmarksflüssigkeit herstellen. Dies ermöglichte es Ionis, ein sehr kompliziertes Computerprogramm zu entwickeln, um vorherzusagen, wie stark die Huntingtin-Senkung im Gehirn und in der Rückenmarksflüssigkeit ist, die durch eine Lumbalpunktion leicht zugänglich ist. Zu diesem Zeitpunkt wurde Ionis von einem großen Pharmapartner, Roche, unterstützt, der über die Ressourcen und Erfahrung verfügt, um komplizierte Humanstudien für ASOs durchzuführen.
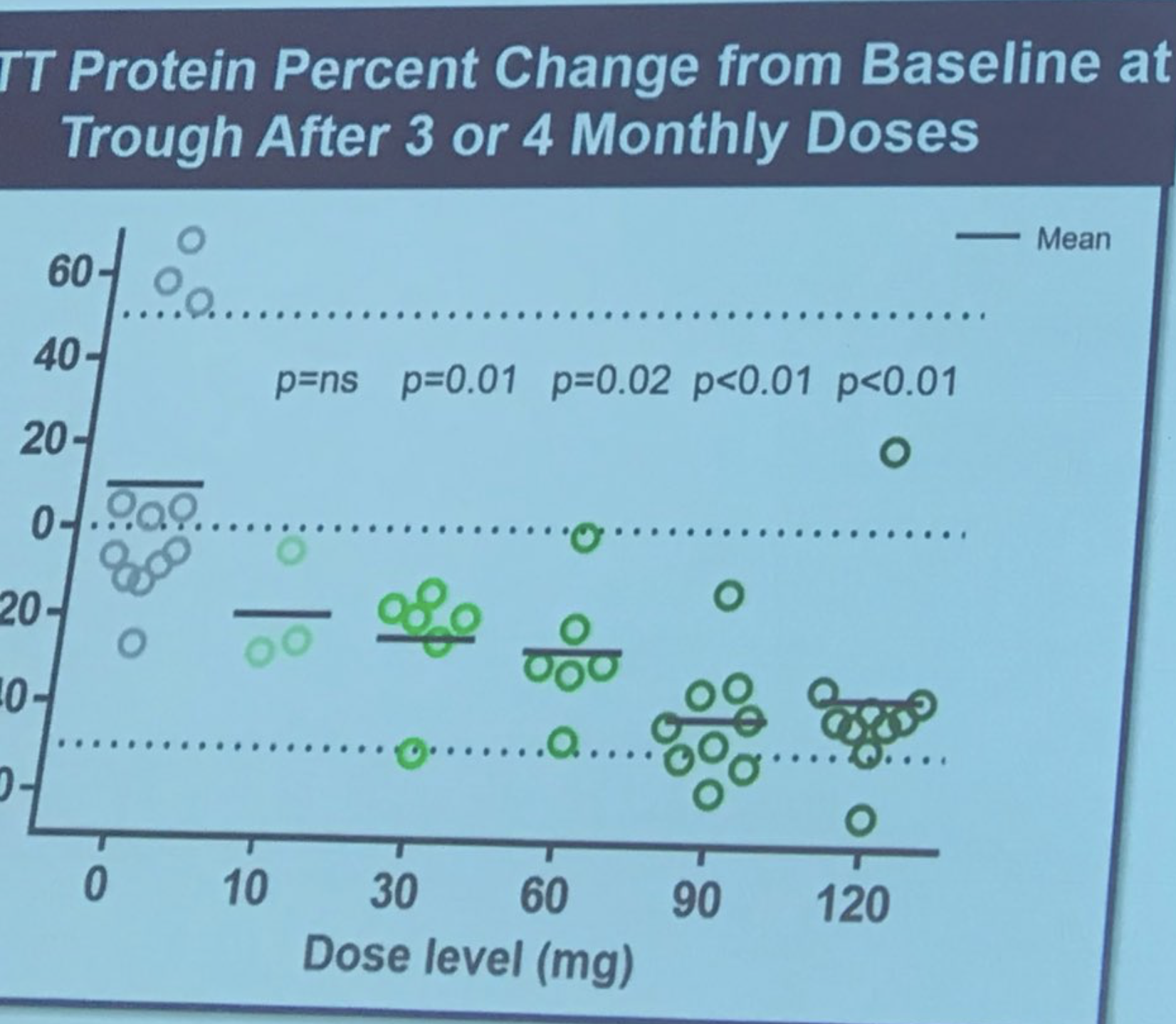
Sarah Tabrizi betritt die Bühne, um die erste Humanstudie der ASO-Behandlung von Ionis/Roche zu beschreiben. Diese Studie war eine „Sicherheitsstudie“ – der Hauptgrund für die Durchführung der Studie war es, festzustellen, ob das Medikament sicher war. Die Studie wurde an 9 Standorten in Großbritannien, Deutschland und Kanada durchgeführt. ASOs wurden Patienten durch Infusion in die Rückenmarksflüssigkeit in einer „eskalierenden Dosis“ verabreicht, was bedeutet, dass frühe Probanden eine niedrige Dosis und spätere Probanden eine höhere Dosis erhielten. Diese sorgfältige Dosissteigerung erfolgt, um Sicherheitsbewertungen durch von der Studie unabhängige Ärzte zu ermöglichen. Diese Studie umfasste 46 unglaublich mutige Freiwillige, die bereit waren, ein gewisses Risiko einzugehen, die ersten Menschen zu sein, die dem Medikament ausgesetzt waren. Forscher konnten die Spiegel des Huntingtin-Proteins in der Rückenmarksflüssigkeit messen – die sie zuvor als sehr gut mit den Hirnspiegeln korreliert gezeigt hatten (die wir, wohlgemerkt, nicht direkt messen können).
Die Größe der Reduktion ist wirklich beeindruckend – im Durchschnitt ganze 40-50 %! Tabrizi beschreibt das Gefühl der Forscher, dass die Huntingtin-Senkung noch bis zu 6 Monate lang weiter verbessert werden könnte. Und hier ist, wie viel Tabrizi voraussagt, dass dies in Bezug auf die Senkung des Hirnproteins entspricht. Ionis hat eine Art Modell entwickelt, das es ihnen ermöglicht, Vorhersagen über die Beziehung zwischen der Huntingtin-Senkung in der Rückenmarksflüssigkeit und in Hirngeweben zu treffen. Dies deutet darauf hin, dass die Huntingtin-Senkung im Hirngewebe recht hoch sein könnte. Die Patienten wurden sehr sorgfältig auf Sicherheit überwacht, es wurden keine größeren unerwünschten Ereignisse festgestellt. Tabrizi: „Das Medikament war sicher und bei allen getesteten Dosen gut verträglich.“ Erfolg! Alle Studienteilnehmer befinden sich nun in einer sogenannten „Open-Label-Verlängerung“ – diejenigen, die Placebo erhielten, wurden auf das Medikament umgestellt und werden weiterhin überwacht. Spontaner Applaus, als Tabrizi den mutigen Freiwilligen der ersten Studie dankt und sie „wahre Forschungshelden“ nennt.
Was für ein Abschluss des Meetings – unglaublich spannende Zeiten liegen vor uns, da Roche und Ionis die nächste Studie planen, die darauf abzielt, festzustellen, ob das Medikament HD-Symptome bei einer größeren Anzahl von Menschen verbessert.
Update: Ionis‘ Community-Statement zu den Ergebnissen.
Mehr erfahren
Weitere Informationen zu unseren Offenlegungsrichtlinien finden Sie in unseren FAQ…


