Eine aktuellere Version dieses Artikels ist verfügbar, wurde aber noch nicht übersetzt.
Wir arbeiten an der Übersetzung des Artikels. Alternativ können Sie ihn in Englisch lesen.
Konferenz über Therapeutika für die Huntington-Krankheit 2025 - Tag 2
HDBuzz berichtete live über Bluesky von der 2025 HD Therapeutics Conference. Lesen Sie die Berichterstattung von Tag 2. #CHDI2025

 Von Dr Sarah Hernandez, Dr Rachel Harding und Dr Tamara Maiuri 1. März 2025 Bearbeitet von Dr Sarah Hernandez und Dr Rachel Harding Übersetzt von Michaela Winkelmann Ursprünglich veröffentlicht am 28. Februar 2025
Von Dr Sarah Hernandez, Dr Rachel Harding und Dr Tamara Maiuri 1. März 2025 Bearbeitet von Dr Sarah Hernandez und Dr Rachel Harding Übersetzt von Michaela Winkelmann Ursprünglich veröffentlicht am 28. Februar 2025
Wir sind zurück für Tag 2 der CHDI Huntington’s Disease Therapeutics Conference! Wir beginnen mit einigen spannenden Vorträgen über genetische Modifikatoren und darüber, wie wir diese in Richtung Therapeutika für die Huntington-Kranhkeit weiterentwickeln können.
Genetische Modifikatoren für die Entwicklung von Medikamenten
In der heutigen Sitzung geht es um genetische Modifikatoren - Gene, die zum Alter des Krankheitsausbruchs beitragen - die durch umfangreiche genetische Studien entdeckt wurden, bei denen die Werte aller Gene bei Personen mit dem Gen für die Huntington-Krankheit untersucht wurden. Auf diese Weise konnten die Forscher Gene identifizieren, die mit einem früheren oder späteren Ausbruch der Huntington-Krankheit korrelieren. Gene, die mit der somatischen Instabilität zusammenhängen, wurden in großen genetischen Studien, den so genannten GWAS (genome wide association studies, genomweite Assoziationsstudien), als Modifikatoren identifiziert. Die Forscher entwickeln GWAS-Daten in Richtung Therapeutika weiter. Dies wäre ohne die Zusammenarbeit zwischen Wissenschaftlern und der Huntington-Gemeinschaft nicht möglich. Super spannend!

Seung Kwak: Es geht nicht nur um somatische Instabilität
Unser erster Redner in dieser Sitzung ist Seung Kwak von CHDI, der über genetische Modifikatoren spricht, die nicht mit der somatischen Instabilität zusammenhängen. Diese Modifikatoren können den Krankheitsbeginn um 7 - 10 Jahre verschieben (das ist viel!), aber sie scheinen die somatische Instabilität nicht zu beeinflussen.
Seung und andere bauen eine Pipeline auf, die ihnen helfen soll, mehr nicht-somatische, mit der Instabilität zusammenhängende Modifikatoren zu identifizieren und herauszufinden, wie sie den Beginn der Symptome beeinflussen. Dies wird dazu beitragen, neue Signalwege bei der Huntington-Krankheit zu identifizieren und die möglichen Moleküle zu diversifizieren, auf die Forscher mit neuen Therapeutika abzielen könnten.
Sobald sie diese Gene identifiziert haben, werden sie versuchen herauszufinden, welche Merkmale der Huntington-Krankheit diese Gene kontrollieren, wie etwa die Geschwindigkeit des Krankheitsverlaufs oder das Auftreten motorischer Symptome. Seung geht davon aus, dass verschiedene Modifikatoren im Verlauf der fortschreitenden Huntington-Krankheit wirken und zu verschiedenen Aspekten des Krankheitsausbruchs beitragen können. Indem wir untersuchen, welche Gene zu welchen Zeitpunkten des Fortschreitens der Huntington-Krankheit beitragen, werden wir eine bessere Vorstellung davon haben, wann wir mit Therapeutika eingreifen können oder sollten, die auf die einzelnen Schritte abzielen.
Seung stellt fest, dass „wir nicht allein sind“, und weist darauf hin, dass andere Krankheiten der Huntington-Krankheit ähnlich sind, wie SCA1, eine andere neurologische Krankheit, die ebenfalls durch expandierende CAGs verursacht wird. Er betont, wie wichtig es ist, von diesen Krankheiten zu lernen, denn das kann dazu beitragen, unser Wissen über die Huntington-Krankheit zu erweitern.
„Interessanterweise scheinen die Nicht-Mismatch-Reparatur-Modifikatoren die Krankheit früher zu beeinflussen. Das heißt, wenn wir einen Weg finden könnten, diese „anderen“ Modifikatoren gezielt zu beeinflussen, könnten wir Wege finden, sehr früh in die Krankheit einzugreifen. “
Indem wir das, was wir über die Huntington-Krankheit wissen, nutzen, um Modifikatoren zu identifizieren, z. B. dass die striatalen Neuronen die am stärksten betroffenen Zellen bei der Huntington-Krankheit sind, können wir die Art und Weise, wie wir Modifikatoren identifizieren, diversifizieren und unsere Erkenntnisse und die Arten von Therapeutika, die wir herstellen könnten, erweitern. Dies könnte dazu beitragen, festzustellen, ob ein kombinatorischer Ansatz am besten geeignet ist.
Marcy MacDonald: Auswirkungen der „anderen“ Modifikatoren
Als Nächstes ist Marcy MacDonald an der Reihe. Sie war ein wichtiges Mitglied des Teams, das 1993 die genetische Mutation identifizierte, die die Huntington-Krankheit verursacht. Sie hat ihre Karriere dem weiteren Verständnis der Huntington-Krankheit gewidmet, um uns einer Behandlung näher zu bringen. Sie wird über die Arbeit ihres Teams zu genetischen Modifikatoren der Huntington-Krankheit berichten. Zunächst geht sie auf GeM-HD ein, eine umfangreiche genetische Studie, in der erstmals einige der genetischen Modifikatoren der Huntington-Krankheit definiert wurden. Marcy erzählte, dass die GeM-HD-Studie ohne die erstaunliche Zusammenarbeit zwischen Huntington-Forschern und der Huntington-Gemeinschaft nicht möglich gewesen wäre.
Sie erinnert uns daran, dass die Huntington-Symptome das Ergebnis komplexer Vorgänge auf molekularer Ebene sind. Wir haben gerade erst damit begonnen, herauszufinden, was das Auftreten der Symptome beeinflusst, und einige Gene sind bereits im Visier! Aber es gibt noch weitere Entdeckungen zu machen, da wir immer mehr Daten erhalten.
Viele der Modifikatoren, von denen wir auf dieser Konferenz bereits gehört haben, sind an der Mismatch-Reparatur beteiligt - einem wichtigen Prozess zur Pflege unserer DNA. Es handelt sich um dieselben Gene, die auch an der somatischen Instabilität beteiligt sind. Es gibt jedoch eine fast ebenso große Zahl von Modifikatoren, die eine völlig andere Biologie haben und die es wirklich wert sind, weiter untersucht zu werden, um herauszufinden, wie sie den Beginn der Huntington-Symptome beeinflussen.
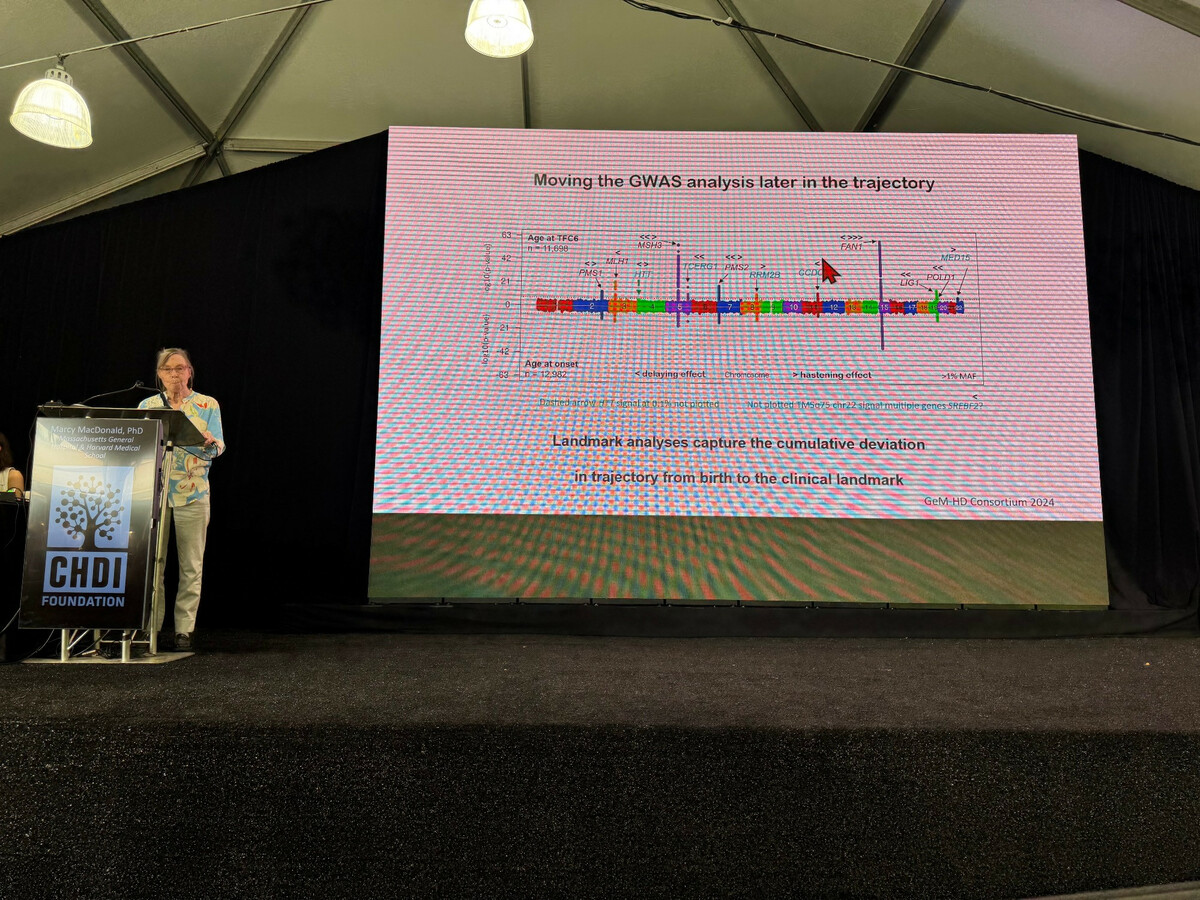
Marcy stellt Daten zur Verfügung, die Modifikatoren in verschiedenen Krankheitsstadien aufzeigen. Interessanterweise scheinen die Nicht-Mismatch-Reparatur-Modifikatoren die Krankheit früher zu beeinflussen. Das bedeutet, wenn wir einen Weg finden könnten, diese „anderen“ Modifikatoren gezielt zu beeinflussen, könnten wir Wege finden, sehr früh in die Krankheit einzugreifen.
Sie verglich auch genetische Modifikatoren in verschiedenen Datensätzen. Es gibt zwar einige Überschneidungen, aber auch einige einzigartige Modifikatoren für jeden Datensatz. Mehrere Datensätze weisen jedoch DNA-Reparaturgene als gemeinsame Treffer auf. Sie betont, dass es wichtig ist, diese Unterschiede zu verstehen. Einige Modifikatoren beeinflussen die Bewegungssymptome der Huntington-Krankheit, während andere sich auf die Denksymptome der Krankheit auszuwirken scheinen. Dies könnte bedeuten, dass Modifikatoren verschiedenen Aspekten der Huntington-Biologie zugrunde liegen.
Die gezielte Beeinflussung dieser aspektspezifischen Modifikatoren könnte den Wissenschaftlern helfen, künftige Behandlungen zu entwickeln, die auf die verschiedenen Arten von Huntington-Symptomen zu unterschiedlichen Zeitpunkten der Krankheit zugeschnitten sind. Dies könnte den Ärzten bei der Huntington-Krankheit die Möglichkeit geben, in Zukunft mit präzisen medizinischen Ansätzen zu behandeln. Marcy schlägt vor, dass es spezifische Modifikatoren für verschiedene biologische Prozesse geben könnte, z. B. für den Beginn der Expansion, die Expansionsrate, zellspezifische Effekte, Zelltoxizität und die Reaktion auf den Verlust von Nervenzellen. Es wäre phantastisch, Ziele für jeden dieser einzigartigen Aspekte der Huntington-Krankheit zu haben!
Außerdem fordert sie die Gemeinschaft auf, nicht nur darüber nachzudenken, welche Zellen bei der Huntington-Krankheit im Laufe der Zeit verloren gehen, sondern auch darüber, welchen Schaltkreisen diese entsprechen. Der Verlust bestimmter Schaltkreise liegt ihrer Meinung nach den verschiedenen Huntington-Symptomen zugrunde.
Jetzt befasst sie sich mit spezifischen Modifikatoren, die nicht die DNA-Reparatur betreffen, und beginnt mit einem dieser Modifikatoren namens Lig1. Es wurden Mäuse hergestellt, die die genetischen Veränderungen in Lig1 aus der GWAS nachahmen, so dass die Forscher eingehend untersuchen können, wie dieses Gen die Huntington-Krankheit beeinflusst.
„Einige Modifikatoren beeinflussen die Bewegungssymptome der Huntington-Krankheit, während andere sich auf die Denksymptome der Krankheit auszuwirken scheinen. Dies könnte bedeuten, dass Modifikatoren verschiedenen Aspekten der Huntington-Biologie zugrunde liegen. “
Ein weiterer Modifikator, den sie erwähnte, ist RRM2B, der mehr bei motorischen Symptomen und weniger bei kognitiven Symptomen eine Rolle spielt. RRM2B trägt dazu bei, dass die Mitochondrien (das Kraftwerk der Zelle) unter Stressbedingungen gesund bleiben. Marcy teilt viele Details über die genauen genetischen Veränderungen mit, die in diesen GWAS gefunden wurden. Sie hebt hervor, dass 12.000 Menschen benötigt wurden, um diese Veränderungen im Zusammenhang mit RRM2B zu erkennen. Sie unterstreicht, wie wichtig es ist, dass Huntinton-Familien zur Forschung beitragen!
Der nächste Modifikator auf Marcys Liste ist die CAA-Sequenz, die manchmal den CAG-Repeat-Abschnitt innerhalb des Huntingtin-Gens unterbricht. Aus der Forschung wissen wir, dass dies der stärkste Modifikator für das Alter des Auftretens der Symptome ist, der das Auftreten von Huntington-Symptomen um bis zu 10 Jahre verzögern kann. Sie hebt hervor, dass die CAA-Unterbrechung die CAG-Instabilität nicht zu beeinflussen scheint, wohl aber die Huntington-Symptome.
Wie macht sie das? Wir wissen es nicht genau. Marcy glaubt, dass es sich indirekt auf die Instabilität auswirkt oder direkt auf bestimmte Arten von Gehirnzellen wirkt und deren Anfälligkeit beeinflusst.
Abschließend fasst Marcy zusammen, dass verschiedene Symptome bei der Huntington-Krankheit zu unterschiedlichen Zeitpunkten auftreten. Genetische Modifikatoren, die in GWAS identifiziert werden, können uns helfen, die Gründe dafür besser zu verstehen und Maßnahmen zu entwickeln, die helfen, die klinischen Anzeichen und Symptome der Huntington-Krankheit zu verändern.
Margaux Hujoel: Somatische Instabilität - Lektionen von 700.000 Menschen

Unsere nächste Referentin ist Margaux Hujoel von der Harvard University. In ihrem Vortrag wird sie darlegen, was sie anhand der genetischen Daten von 700.000 Menschen, die Proben wie Blut oder Rückenmarksflüssigkeit für die Forschung gespendet haben, über die Ursachen und Folgen der somatischen Instabilität gelernt hat. Um genetische Variationen bei der Huntington-Krankheit und anderen Krankheiten zu verstehen, benötigen wir umfangreiche Datensätze von Tausenden von Menschen, um uns der Ergebnisse sicher sein zu können. Da sich die Genomsequenzierungstechnologien in den letzten Jahrzehnten dramatisch weiterentwickelt haben, haben wir jetzt Zugang zu riesigen Datensätzen - sehr spannend.
Sie beginnt mit einer Zusammenfassung des Konzepts, dass die Huntington-Krankheit durch somatische Instabilität, d. h. die ständige Ausdehnung der krankheitsverursachenden CAG-Wiederholung, verursacht wird. Allerdings wissen die Forscher noch nicht genau, warum die Instabilität bei der Huntington-Krankheit so wichtig ist.
Margaux verlässt das Huntingtin-Gen und untersucht, wie somatische Instabilität im gesamten genetischen Code (Genom) auftritt, um zu sehen, welche Lehren aus einem breiteren Blickwinkel gezogen werden können. Andere Krankheiten werden durch erweiterte Wiederholungen verursacht, so dass wir mehr über die Huntington-Krankheit lernen könnten, wenn wir sie untersuchen.
Zwei dieser Krankheiten sind die Myotonische Dystrophie und die Fuchsche Hornhautdystrophie, eine Augenkrankheit. Die Erforschung dieser Krankheiten zeigt, dass neue Fälle durch eine Instabilität der Wiederholungen entstehen, die eine sich wiederholende DNA-Sequenz auf eine Länge bringt, die eine Krankheit verursacht, ähnlich wie bei der Huntington-Krankheit.
Auf der ganzen Welt gibt es verschiedene Biobanken, in denen Gewebe und Flüssigkeiten gesammelt werden, die von Menschen mit Krankheiten gespendet wurden. Anhand von Proben aus diesen Biobanken lernen Margaux und ihr Team mehr über die somatische Instabilität, die für alle Krankheiten von Bedeutung ist.
„Es gibt 18 verschiedene Stellen im menschlichen Genom, die für somatische CAG-Instabilität empfindlich sind, von denen 9 als krankheitsverursachend bekannt sind. “
Die Analyse langer Wiederholungen in der DNA ist mit einigen technischen Herausforderungen verbunden, aber Margauxs Team hat eine Lösung gefunden und herausgefunden, dass die überwiegende Mehrheit der Erweiterungen in nur einer Handvoll von Genen vorkommt, was dazu beiträgt, die Bereiche einzugrenzen, auf die wir uns konzentrieren sollten.
Es gibt 18 verschiedene Stellen im menschlichen Genom, die für somatische CAG-Instabilität empfindlich sind, von denen 9 als krankheitsverursachend bekannt sind. Anhand von Proben in der Biobank von verwandten Personen können Margaux und ihr Team genetische Veränderungen im Genom über mehrere Generationen hinweg kartieren. Sie fand heraus, dass CAG-Expansionen häufiger expandieren als kontrahieren und dass Expansionen häufiger bei größeren CAG-Wiederholungslängen auftreten. Dies ist für Huntington-Forscher nichts Neues, aber es ist für uns interessant zu wissen, dass dieses Phänomen nicht nur bei der Huntington-Krankheit auftritt, sondern im gesamten Genom.
Die Forscher untersuchten auch, wie sich die Expansionen zwischen verschiedenen Gewebetypen unterscheiden, z. B. zwischen Blut und Hirngewebe. Dies ist insofern von Bedeutung, als wir wissen müssen, welche Bioflüssigkeiten oder Gewebe am besten geeignet sind, um die Ausdehnung zu verfolgen, und um Veränderungen der Ausdehnung in bevorstehenden klinischen Studien zu messen, die darauf abzielen, die Ausdehnung zu verlangsamen.
Margaux zeigte Daten für verschiedene Krankheiten, bei denen die wiederholten Ausdehnungen in der Keimbahn (Ei- und Samenzellen) wahrscheinlicher waren als im Blut, und umgekehrt. Dies deutet darauf hin, dass die zelltypspezifischen Unterschiede bei CAG-Repeat-Krankheiten nicht unbedingt gleich sind, ABER die Zelltypspezifität scheint ein gemeinsames Merkmal zu sein.
Ein weiteres gemeinsames Merkmal dieser Krankheiten ist, dass ähnliche Gene zur Repeat-Instabilität beitragen, z. B. Modifikatoren, die mit der DNA-Reparatur zusammenhängen, wie MSH3, PMS2 und FAN1 - alles Gene, die bei der Huntington-Krankheit wegen ihrer Rolle bei der somatischen Instabilität intensiv untersucht werden.

Margaux schlägt vor, dass wir einige ihrer Forschungsergebnisse auf die Huntington-Krankheit anwenden können. Sie gibt zu bedenken, dass die somatische Expansion im Blut vielleicht nicht mit dem übereinstimmt, was im Gehirn vor sich geht, dass sie aber dennoch ein interessanter Biomarker für Therapeutika sein könnte, die auf die Kontrolle der Expansion abzielen. Die Forschung arbeitet hart daran, Biomarker zu finden, mit denen die somatische Expansion verfolgt werden kann, während potenzielle Behandlungen auf dem Weg in die Klinik sind. Allerdings können wir während der klinischen Studien keine Gehirnproben entnehmen, so dass Blut eine Möglichkeit sein könnte, um festzustellen, ob solche Behandlungen die gewünschte Wirkung haben.
Sollte sich herausstellen, dass Blutproben kein gutes Surrogat für solche Therapeutika sind, müssen wir unsere Strategie möglicherweise überdenken. Dies stellt die derzeitigen Ansätze in der Huntington-Forschung in Frage, aber genau darum geht es ja bei Konferenzen! Es geht darum, das, was wir wissen, in Frage zu stellen, die Menschen dazu zu bringen, die Dinge auf andere Weise zu betrachten und die Huntington-Forschung mit einer breiten Perspektive voranzubringen.
Aaron Gitler: Modifikatoren - Lektionen aus anderen Krankheiten
Als nächstes wird Aaron Gitler, der sich mit der ALS (Lou-Gehrig-Krankheit) befasst, über Erkenntnisse aus seiner eigenen Arbeit berichten, die seiner Meinung nach für die Huntington-Krankheit relevant sein könnten. Konkret geht es dabei um seine Arbeit über genetische Modifikatoren.
ALS kann durch Veränderungen an einem Gen namens TDP-43 verursacht werden. Wie Huntingtin kann auch dieses Gen die mit der Krankheit verbundenen Proteinverklumpungen hervorrufen. Interessanterweise wurde dieses Gen vor kurzem auch bei der Huntington-Krankheit entdeckt.
„Dies stellt die derzeitigen Ansätze in der Huntington-Forschung in Frage, aber genau darum geht es ja bei Konferenzen! Es geht darum, das, was wir wissen, in Frage zu stellen, die Menschen dazu zu bringen, die Dinge auf andere Weise zu betrachten und die Huntington-Forschung mit einer breiten Perspektive voranzubringen. “
Aaron fand ein Gen mit der Bezeichnung ATXN2, das die Proteinverklumpung von TDP-43 unterdrückt. ATXN2 scheint die ALS zu beeinflussen, verursacht aber auch eine andere Krankheit, die spinozerebelläre Ataxie 2. Er fand heraus, dass in einigen ALS-Fällen eine genetische Erweiterung der CAG-Wiederholungen im ATXN2-Gen vorliegt. Interessanterweise hat er herausgefunden, dass unterschiedliche Längen der CAG-Wiederholungen in ATXN2 unterschiedliche Krankheitsmerkmale in verschiedenen Zellen verursachen. Ein ziemlich komplexes System!
Wenn Aaron bei Mäusen, die ein Modell für ALS darstellen, die ATXN2-Konzentration reduziert, leben die Mäuse viel länger und die Krankheitsmerkmale in den Gehirnzellen scheinen zu verschwinden. Dies deutet darauf hin, dass ATXN2 ein gutes Ziel für ALS-Therapeutika sein könnte.
Seine Arbeit deutet darauf hin, dass bei Menschen mit einer Krankheit Teile der genetischen Information in Proteinen enthalten sind, die bei Menschen ohne diese Krankheit nicht vorhanden sind. Die Aufnahme oder der Ausschluss dieser genetischen Informationen erfolgt durch einen Prozess, der Spleißen genannt wird.
Durch diese Arbeit hat er möglicherweise eine genetische Ursache für die TDP-43-Krankheit identifiziert, die therapeutisch genutzt werden könnte. Er vermutet, dass bei der Huntington-Krankheit ähnliche biologische Mechanismen im Spiel sein könnten, insbesondere angesichts der kürzlich veröffentlichten Assoziation zwischen Huntington und TDP-43.
Julien Marnet: Auf der Jagd nach dem Hauptschalter bei der Huntington-Krankheit
Unser letzter Redner des Tages ist Julien Mamet, der bei Core Biotherapeutics arbeitet, einem Unternehmen, das sich auf die Entwicklung von Therapeutika spezialisiert hat, die auf Gene abzielen, die als „Transkriptionsfaktoren“ bezeichnet werden - Gene, die wie Hauptregulatoren wirken und das Niveau vieler anderer Gene steuern.
Julien untersucht große Datensätze und kartiert, wie Gene innerhalb bestimmter Zelltypen hierarchisch miteinander verbunden sind und sich gegenseitig regulieren. Indem er dies in Zellen mit und ohne Huntington-Krankheit tut, kann er Unterschiede feststellen und herausfinden, wie man die Hauptregulatoren in diesen hierarchischen Netzwerken angreifen kann. Julien erinnert uns daran, dass nicht alle Transkriptionsfaktoren gleich sind, weshalb viel Aufwand betrieben wird, um zu verstehen, welche dieser Hauptregulatoren dominant sein könnten. Sie bezeichnen diese als die „Kern“-Komponenten des Netzwerks. Bei Krankheiten wird angenommen, dass diese „zentralen“ Hauptregulatorgene die Krankheit auslösen.
Sie arbeiten daran, viele verschiedene Datensätze zu integrieren, um eine Bibliothek von Netzwerken zu erstellen und Kerne innerhalb dieser Netzwerke zu identifizieren. Dies wird ihnen helfen, Targets zu identifizieren, gegen die sie Therapeutika entwickeln können, die ihrer Meinung nach die Krankheitszeichen und -symptome verbessern könnten. Bei der Huntington-Krankheit beginnen sie, diese Netzwerke anhand von Datensätzen verschiedener Zelltypen im Gehirn zu erstellen. Aus diesen Netzwerken haben sie Kerngene mit der Bezeichnung „HOX“ identifiziert. HOX-Gene sind besonders stark in Neuronen, die bei der Huntington-Krankheit anfällig sind.
Bei der Huntington-Krankheit scheinen diese HOX-Gene Tausende von Genen zu verändern, die für die ordnungsgemäße Funktion der Gehirnzellen notwendig sind. Julien stellt fest, dass diese HOX-Gene zentrale Gene in den Netzwerken der frühen und späten Stadien der Huntington-Krankheit sind. Julien schlägt vor, dass die HOX-Gene in Gehirnzellen, die nicht stark von der Huntington-Krankheit betroffen sind, unverändert bleiben, so dass sie sicher für Therapien geeignet sein sollten. Interessanterweise sehen sie etwas Ähnliches bei anderen Krankheiten, die durch CAG-Wiederholungen verursacht werden, was darauf hindeutet, dass eine mögliche Therapie, die auf HOX abzielt, nicht nur bei der Huntington-Krankheit wirksam sein könnte.
Das war’s für heute! Bleiben Sie dran und erfahren Sie am letzten Tag der Therapeutics Conference weitere spannende Neuigkeiten aus der Forschung zur Huntington-Krankheit!


