
Konferenz über Therapeutika für die Huntington-Krankheit 2025 – Tag 1
HDBuzz berichtete live über Bluesky von der 2025 HD Therapeutics Conference. Lesen Sie die Berichterstattung von Tag 1. #CHDI2025
Hallo aus Palm Springs! Das HDBuzz-Team ist hier und bereit, über all die spannenden wissenschaftlichen Erkenntnisse zu berichten, die wir in den nächsten drei Tagen von Huntington-Experten hören werden, die aus der ganzen Welt angereist sind, um an der 20. jährlichen CHDI-Konferenz über Therapeutika für die Huntington-Krankheit teilzunehmen. Machen Sie sich bereit, in den nächsten 3 Tagen einige spannende Forschungsergebnisse zur Huntington-Krankheit zu verfolgen!
Updates zu klinischen Studien
Unsere erste Vortragsreihe sind Updates von Pharmaunternehmen, die laufende klinische Studien durchführen.

Image credit: Jeff Carroll
PTC Therapeutics – PIVOT-HD testet Votoplam
Den Anfang macht Amy-Lee Breadlau von PTC Therapeutics mit einem Update zu Votoplam, ehemals PTC-518. Votoplam ist ein HTT-senkendes Medikament, das in Tablettenform eingenommen wird. Die Idee ist, dass durch die Senkung des krankheitsverursachenden Proteins die Anzeichen und Symptome der Huntington-Krankheit verringert werden.
Amy-Lee teilte zunächst die fantastische Nachricht mit, dass PTC eine aufregende neue Zusammenarbeit mit Novartis, einem großen Arzneimittelhersteller, eingegangen ist. PTC wird die laufende PIVOT-HD-Studie durchführen, aber Novartis wird für alle künftigen klinischen Studien, wie die geplante Phase 3, verantwortlich sein.
Bislang haben die Teilnehmer der PIVOT-HD-Studie Votoplam 12 Monate lang eingenommen. Die Forscher wollten vor allem wissen, ob sich die BiomarkerBiomarker Irgendeine Art von Test – inklusive Bluttest, Gedächtnistest und Gehirnscan – der das Fortschreiten einer Krankheit wie der Huntington-Krankheit messen oder vorhersagen kann. Biomarker können klinische Studien von neuen Medikamenten schneller und verlässlicher machen. – biologische Messgrößen, die das Fortschreiten der Huntington-Krankheit anzeigen – bei Personen, die das Medikament so lange einnehmen, positiv verändern. Der zuverlässigste BiomarkerBiomarker Irgendeine Art von Test – inklusive Bluttest, Gedächtnistest und Gehirnscan – der das Fortschreiten einer Krankheit wie der Huntington-Krankheit messen oder vorhersagen kann. Biomarker können klinische Studien von neuen Medikamenten schneller und verlässlicher machen., den wir derzeit haben, ist Neurofilament Light (NfLNfL Biomarker für die Gesundheit des Gehirns). Wir wissen, dass die NfLNfL Biomarker für die Gesundheit des Gehirns-Werte mit fortschreitender Huntington-Erkrankung ansteigen. Spannenderweise zeigt Amy, dass die NfLNfL Biomarker für die Gesundheit des Gehirns-Werte bei Menschen, die 12 Monate lang Votoplam erhalten haben, konstant bleiben.
Jetzt zeigt sie sehr interessante Daten, die darauf hindeuten, dass sich die klinischen Messwerte bei Menschen, die das Medikament seit einem Jahr einnehmen, verbessern. Letzten Endes ist das genau das, was wir wollen! Ein Medikament, das die klinischen Anzeichen von der Huntington-Krankheit verbessert, ist ein Medikament, das gegen die Huntington-Krankheit wirkt!
Die endgültigen Ergebnisse von PIVOT-HD werden voraussichtlich in diesem Sommer veröffentlicht. Wir werden Sie natürlich auf dem Laufenden halten, sobald wir mehr erfahren!
Roche – GENERATION-HD2 untersucht Tominersen
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. nächster Redner berichtet Peter McColgan von Roche über den neuesten Stand ihres Portfolios für die Huntington-Krankheit. Die größte Studie war GENERATION-HD1 für das HTT-senkende Medikament Tominersen. Diese Studie war zwar nicht erfolgreich, lieferte aber die Daten für die laufende GENERATION-HD2-Studie, in der Tominersen an einer spezifischeren Gruppe von Menschen mit der Huntington-Krankheit getestet wird.
Die große Neuigkeit, die Peter heute mitteilt, ist, dass die GENERATION-HD2-Studie vollständig rekrutiert ist. Sie sind in 15 Ländern an 70 Standorten aktiv und hoffen, die Studie bis Ende des Jahres abschließen zu können.
Roche arbeitet aktiv mit dem HD Regulatory Science Consortium zusammen, um die gesammelten Daten weiterzugeben. Roche stellt auch Daten über den natürlichen Verlauf und Daten aus der GENERATION-HD1-Studie von Personen zur Verfügung, die nicht mit dem Medikament behandelt wurden. Diese Art von Daten ermöglicht es den Forschern zu verstehen, wie die Huntington-Krankheit normalerweise im Alltag und im Alter fortschreitet. Diese wertvollen Informationen werden den Forschern zur Verfügung stehen und uns helfen, bessere Studien zu entwickeln.
Roche arbeitet auch mit CHDI zusammen, um den Biomarker NfL besser zu verstehen. Ihr Ziel ist es, herauszufinden, ob NfLNfL Biomarker für die Gesundheit des Gehirns mehr alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. ein BiomarkerBiomarker Irgendeine Art von Test – inklusive Bluttest, Gedächtnistest und Gehirnscan – der das Fortschreiten einer Krankheit wie der Huntington-Krankheit messen oder vorhersagen kann. Biomarker können klinische Studien von neuen Medikamenten schneller und verlässlicher machen. ist und alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. diagnostischer Test für die Huntington-Krankheit verwendet werden könnte. Die Verwendung von NfLNfL Biomarker für die Gesundheit des Gehirns alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. diagnostischer Test könnte uns Aufschluss darüber geben, wo sich die Menschen in der Progression der Krankheit befinden. Dies könnte dazu beitragen, künftige Medikamente maßzuschneidern und die Pflegepläne auf das jeweilige Krankheitsstadium abzustimmen.
„An alle, die an diesen Beobachtungsstudien teilnehmen – DANKE! SIE verändern wirklich das Gesicht der Huntington-Forschung! Dank Ihres Beitrags erfahren die Huntington-Wissenschaftler jeden Tag mehr über diese Krankheit und erhalten Antworten auf Fragen, die uns zu einer Behandlung verhelfen werden.“
Die Forscher verwenden mehrere Datensätze von Enroll-HD, HD-Clarity, Track-HD und Track-On, um zu sehen, wie sich NfLNfL Biomarker für die Gesundheit des Gehirns im Laufe der Zeit verändert. Zusammengenommen liefern diese Datensätze Proben von fast 7.000 Menschen, die mit der Huntington-Krankheit leben!
Wir können gar nicht hoch genug einschätzen, wie wichtig diese Datensätze für die Huntington-Wissenschaftlergemeinschaft sind. Also an alle, die an diesen Beobachtungsstudien teilnehmen – DANKE! Sie verändern wirklich das Gesicht der Huntington-Forschung! Dank Ihres Beitrags erfahren die Huntington-Wissenschaftler jeden Tag mehr über diese Krankheit und erhalten Antworten auf Fragen, die uns zu einer Behandlung verhelfen werden. Wenn Sie mehr erfahren oder einen Beitrag zur Huntington-Forschung leisten möchten, können Sie die Enroll-HD-Website besuchen.
Wave Life Sciences – SELECT-HD untersucht WVE-003
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Nächstes folgt ein Update von Wave Life Sciences zu ihrem HTT-senkenden Medikament WVE-003, das in ihrer Studie SELECT-HD getestet wird. Dieses Medikament ist einzigartig, weil es speziell auf die erweiterte HTT-Kopie abzielt, die die Huntington-Krankheit verursacht. Die gezielte Ausrichtung auf die erweiterte HTT-Kopie hat Vorteile.
Wave glaubt auch, dass die Ausrichtung auf die erweiterte Kopie einen Einfluss auf die somatische Instabilität haben wird, die fortwährende Ausdehnung der CAG-Wiederholung innerhalb des HTT-Gens. Das wäre zwar super cool, aber wir haben noch keine Daten, die zeigen, dass WVE-003 die somatische Instabilität tatsächlich beeinflussen kann.
Wave hat gezeigt, dass sich die Hirnatrophie bei Menschen, die WVE-003 einnehmen, verlangsamt. Das Unternehmen hofft, dieses Maß in künftigen klinischen Studien alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Indikator für die klinischen Ergebnisse verwenden zu können.
Allerdings müssen wir alle Daten zur Hirnatrophie im Zusammenhang mit den Biomarkern sorgfältig interpretieren. Die Betrachtung der Hirnatrophie allein kann uns nicht wirklich sagen, ob sich die Huntington-Krankheit verbessert, da hier auch andere Faktoren eine Rolle spielen könnten, wie z. B. die Schwellung des Gehirns. Wenn ein Medikament eine EntzündungEntzündung Aktivierung des Immunsystems, vermutlich am Huntington-Krankheitsprozess beteiligt des Gehirns hervorruft, könnte dies den Anschein erwecken, dass das Gehirn weniger schrumpft, was aber nicht unbedingt bedeutet, dass das Medikament eine positive Wirkung hat.
Wenn es aber auch BiomarkerBiomarker Irgendeine Art von Test – inklusive Bluttest, Gedächtnistest und Gehirnscan – der das Fortschreiten einer Krankheit wie der Huntington-Krankheit messen oder vorhersagen kann. Biomarker können klinische Studien von neuen Medikamenten schneller und verlässlicher machen. gibt, die darauf hindeuten, dass das Medikament die biologischen Messwerte der Huntington-Krankheit verbessert, wäre das eine fantastische Sache! Wir müssen alle Daten zur Hirnatrophie sorgfältig interpretieren, wenn wir mehr erfahren und wenn WVE-003 die klinische Studienpipeline durchläuft. Wave hofft, bis Ende dieses Jahres mit einer Phase-2/3-Studie voranzukommen. Wir werden Sie auf dem Laufenden halten, sobald wir mehr erfahren!
UniQure – Untersuchung von AMT-130
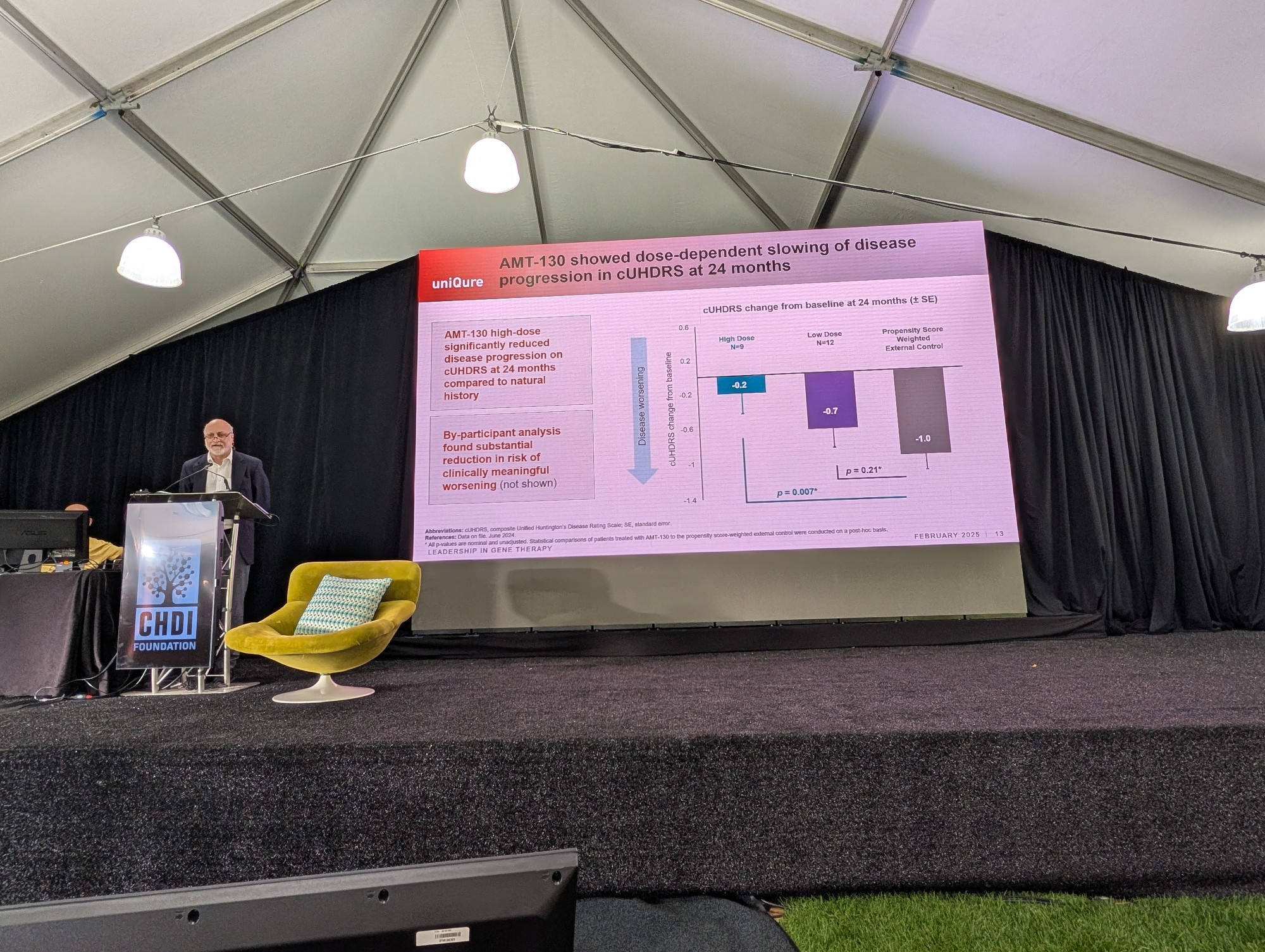
Unser letzter Redner in dieser Sitzung ist David Margolin von uniQure, der über den neuesten Stand von AMT-130 berichtet, einer HTT-senkenden Gentherapie, die durch eine Gehirnoperation verabreicht wird. Sein Hauptaugenmerk liegt heute auf der kürzlich erfolgten Abstimmung mit der FDA auf dem Weg zu einer beschleunigten Zulassung für AMT-130.
AMT-130 ist ein Medikament, das durch eine Gehirnoperation verabreicht wird und sowohl die normale alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. auch die toxische Form von Huntingtin senkt. Das Medikament setzt direkt am Anfang des Huntingtin-Botschaftsmoleküls an, was bedeutet, dass auch die toxische Fragmentform von Huntingtin gesenkt werden soll.

UniQure hat sich bei der FDA um die RMAT – Regenerative Medicine Advanced Therapy Designation – beworben. Dieser Antrag war erfolgreich! Dies ist wichtig, weil sich dadurch die Zeit, die AMT-130 bis zur Markteinführung benötigen könnte, um mehrere Jahre verkürzt. Dies ist natürlich für alle Huntington-Familien sehr wichtig! UniQure führt weiterhin Gespräche mit der FDA über die genauen Daten, die sie für eine beschleunigte Zulassung ihres Medikaments benötigen.
UniQure plant, Daten zum natürlichen Verlauf zu verwenden, um herauszufinden, wie gut ihr Medikament wirkt. Das bedeutet, dass sie die Teilnehmer ihrer Studie mit dem vergleichen werden, was im Durchschnitt für Menschen mit der Huntington-Krankheit zu erwarten ist, die das gleiche Alter haben und so weiter, die aber das Medikament nicht erhalten haben. Dies unterscheidet sich ein wenig von einer PlaceboPlacebo Ein Placebo ist ein Scheinmedikament, das keine Wirkstoffe enthält. Der Placeboeffekt ist ein psychologischer Effekt, der verursacht, dass sich Menschen besser fühlen, auch wenn sie eine Tablette einnehmen, die nicht wirkt.-Kontrolle, wie sie von den Unternehmen üblicherweise verwendet wird.
Einer der wichtigsten Punkte, auf die sich die FDA mit uniQure geeinigt hat, ist die Verwendung von cUHDRS alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Maßstab für die WirksamkeitWirksamkeit Ein Maßstab, ob eine Therapie wirkt. des Medikaments. cUHDRS ist eine Kombination aus vielen verschiedenen Messwerten für alle Arten von Anzeichen und Symptomen der Huntington-Krankheit. Sie gilt alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. besser alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. eine einzige Messung, da jede einzelne mit verschiedenen Vorbehalten behaftet ist. Bei kombinierter Anwendung können diese Vorbehalte jedoch ausgemerzt werden, und wir können recht schnell feststellen, ob das Medikament WIRKLICH wirkt.
Außerdem stimmte die FDA zu, die NfLNfL Biomarker für die Gesundheit des Gehirns-Werte in der Rückenmarksflüssigkeit alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. unterstützenden Beweis für die WirksamkeitWirksamkeit Ein Maßstab, ob eine Therapie wirkt. von AMT-130 zu betrachten. NfLNfL Biomarker für die Gesundheit des Gehirns steigt bei Menschen mit der Huntington-Krankheit typischerweise im Laufe der Zeit an. Wenn also die NfLNfL Biomarker für die Gesundheit des Gehirns-Werte sinken oder konstant bleiben, ist das eine gute Nachricht für die Gesundheit des Gehirns von Menschen mit der Huntington-Krankheit.
Die fantastische Nachricht ist, dass sich die FDA und die Hersteller von Huntington-Medikamenten nun darauf geeinigt haben, welche Kriterien und Maßnahmen zu erwarten sind, um nachzuweisen, dass ein Medikament gut genug wirkt und sicher genug ist, damit die FDA es zulassen kann.
Connecting The Dots: HTT-Biologie vom Labor in die Klinik
Unsere nächste Sitzung befasst sich mit den Erkenntnissen, die wir im Labor über die Biologie der Huntington-Krankheit gewonnen haben und die in klinische Studien einfließen.
Longzhi Tan: Genetische Architektur
Den Anfang macht Longzhi Tan von der Stanford University, der über seine Arbeit zur Architektur des genetischen Materials berichtet und darüber, wie die Huntington-Krankheit die Form und den Sitz des genetischen Materials in der Zelle beeinflusst. Genetische Architektur klingt super cool! Aber was genau ist das?
Wir haben eine Menge genetisches Material, das uns alle einzigartig macht, und es ist wichtig, wo es in der Zelle sitzt. Die DNA in jeder Zelle unseres Körpers ist 2 Meter lang! Damit sie in jede winzige Zelle passt, muss sie sich falten und zusammendrücken lassen, um eingequetscht zu werden. Tan ist daran interessiert, genau zu untersuchen, wie die DNA in den Zellen organisiert ist und wo jedes Gen zu finden ist.
Erstaunlicherweise kann Tan verschiedene Zelltypen allein dadurch bestimmen, wo sich die DNA in der Zelle befindet. Wie die DNA organisiert ist und wo sie sich in der Zelle befindet, ändert sich im Laufe des Lebens.
„Die fantastische Nachricht ist, dass wir jetzt eine Vereinbarung zwischen der FDA und den Huntington-Arzneimittelherstellern darüber haben, welche Kriterien und Maßnahmen zu erwarten sind, um zu zeigen, dass ein Medikament gut genug funktioniert und sicher genug ist, damit die FDA ein Medikament zulassen kann. “
Jetzt untersucht er, wie sich die Huntington-Krankheit auf die Architektur des genetischen Materials in Mäusen auswirkt, die alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Modell für die Krankheit dienen. Er zeigt dem Publikum die allererste 3D-Karte, die zeigt, wie das genetische Material in Zellen von Huntington-Mäusen aussieht. Die Huntington-Krankheit verursacht drastische Veränderungen, insbesondere in Zellen, die bei der Huntington-Krankheit anfällig sind. Tan erklärt, dass er glaubt, dass diese Veränderungen zu einem Verlust der „Zellidentität“ führen – also zu Genen, die bestimmte Zelltypen zu dem machen, was sie sind.
Tan untersucht auch, wie Gene, die die somatische Instabilität kontrollieren, die genetische Architektur beeinflussen könnten. Er untersuchte insbesondere das Modifikatorgen Msh3. Wenn der Msh3-Spiegel in Mäusen mit der Huntington-Krankheit gesenkt wird, scheint dies die durch die Huntington-Krankheit verursachten Veränderungen der genetischen Architektur zu korrigieren. Sehr schick!
Die Botschaft lautet, dass die gezielte Beeinflussung von Msh3 in verschiedenen Huntington-Modellen dazu beitragen könnte, viele der Merkmale der Huntington-Krankheit wieder zu normalisieren. Diese Art von wirklich detaillierter Analyse spricht dafür, dass Msh3 ein gutes Ziel für Wissenschaftler ist, die versuchen, neue Medikamente zu entwickeln.
Kejia Wu: Vom Computer entworfene Proteine
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. nächstes ist Kejia Wu von der University of Washington in Seattle an der Reihe. Sie hat kürzlich im Labor von David Baker, dem Nobelpreisträger von 2024, promoviert. In ihrer Forschung versucht sie, neue Arten von Proteinen für bestimmte Aufgaben zu entwickeln. Diese Arbeit nutzt alle Arten von spezialisierten Deep-Learning- und KI-Guides.
Kejia interessiert sich für die schlaffen Teile von Proteinmolekülen. Es hat sich herausgestellt, dass diese schlaffen Bereiche wirklich wichtig für alle Arten von Biologie sind, aber traditionell alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. „nicht beeinflussbar“ galten. Sie hofft, diese schlaffen Bereiche mit neu entworfenen Proteinen, die ihre KI-gesteuerten Computermethoden halluzinieren, gezielt angreifen zu können.
Das Huntingtin-ProteinHuntingtin-Protein Das Protein, das durch das Huntington-Gen hergestellt wird. hat viele dieser schlaffen Regionen, von denen Huntington-Wissenschaftler gezeigt haben, dass sie wichtig dafür sind, wie das Protein weiß, wo es in der Zelle hingehört und mit welchen Proteinen es zusammenarbeiten muss. Kejia wendet ihre Technologie auf Huntingtin an, um neue Proteine zu entwickeln, die auf die lange Glutaminkette am Anfang des krankheitsverursachenden Proteins abzielen. Im normalen Huntingtin-ProteinHuntingtin-Protein Das Protein, das durch das Huntington-Gen hergestellt wird. gibt es eine ähnliche Kette von Glutaminen, aber sie ist viel kürzer. Der Unterschied ist jedoch sehr subtil, so dass es schwierig ist, etwas Selektives zu finden!
Die Huntington-Gemeinschaft kann sich glücklich schätzen, so viele Technologieexperten wie Kejia und Tan zu haben, die daran interessiert sind, die Huntington-Krankheit zu verstehen und uns dabei zu helfen, neue Wege zu finden, wie wir Medikamente für die Zukunft entwickeln können!
Gill Bates: Toxisches Fragment HTT1a
Unser nächster Redner ist Gill Bates vom University College London. Gills Gruppe konzentriert sich auf eine Art von Huntingtin-ProteinHuntingtin-Protein Das Protein, das durch das Huntington-Gen hergestellt wird. namens HTT1a. Dabei handelt es sich um ein kleines Fragment des Huntingtin-Proteins, das in Zellen toxische Klumpen bildet.
Gills Team untersucht, wie viel von der HTT1a-Form bei verschiedenen Huntington-Modellen gebildet wird. Es zeigt sich, dass je länger die Wiederholungslänge der Huntington-Mutation ist, desto mehr HTT1a gebildet wird. Gleichzeitig nimmt die Menge der Huntingtin-Form in voller Länge ab.
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Nächstes untersuchte das Team von Gill, ob sich die Menge des Proteins, das toxische Klumpen bildet, verändert. Sie glauben, dass HTT1a der Katalysator sein könnte, der die Verklumpung des Huntingtin-Proteins in Gang setzt. Um dies herauszufinden, verwendeten sie ein spezielles Mausmodell der Huntington-Krankheit, das kein HTT1a bilden kann. Diese Mäuse haben viel weniger Klumpen alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. normale Huntington-Mäuse, und diese Klumpen bilden sich langsamer.
Es wird angenommen, dass diese Proteinverklumpungen, auch AggregateAggregate Klumpen von Proteinen, die sich innerhalb von Zellen bei der Huntington-Krankheit und anderen degenerativen Erkrankungen bilden genannt, auf verschiedene Weise toxisch für Zellen sind. Eine der am besten untersuchten Methoden ist die Art und Weise, wie Verklumpungen in den Zellkernen, wo das genetische Material gespeichert ist, Auswirkungen darauf haben können, welche Gene ein- oder ausgeschaltet werden. Gill und ihr Team untersuchten, wie Klumpen im ZellkernZellkern Ein Teil der Zelle, der die Gene enthält (DNA) mit diesen genetischen Veränderungen zusammenhingen. Je mehr Klumpen sie sahen, desto mehr genetische Veränderungen sahen sie.
Die Huntington-Wissenschaftler versuchen immer noch herauszufinden, inwieweit jeder Typ des Huntingtin-Proteins für die Entstehung der Krankheit bei der Huntington-Krankheit verantwortlich ist. Gill und Kollegen verwenden genetische Instrumente, die nur auf einen Typ des Huntingtin-Proteins abzielen, um herauszufinden, was vor sich geht.
Bei der Betrachtung der Aggregatspiegel und der Frage, welche Gene ein- und ausgeschaltet werden, stellten sie fest, dass die Senkung der HTT1a-Menge und des erweiterten Huntingtins den größten Effekt hatte, während die Senkung anderer Huntingtin-Typen in den von ihnen verwendeten Mausmodellen keine große Wirkung zeigte. Dies ist für die Forschung von Bedeutung, da es in der Klinik alle möglichen Therapien zur Senkung des Huntingtin-Spiegels gibt, die alle leicht unterschiedlich wirken und jeweils verschiedene Formen des Huntingtin-Proteins in der Zelle senken. Wir wissen noch nicht, welche davon bei Menschen am besten wirkt. Je mehr wir also auf dieser detaillierten molekularen Ebene verstehen, desto besser.
Gill arbeitet mit dem Khvorova-Labor in den USA zusammen, um neue Instrumente zur Senkung der HTT1a-Konzentration zu entwickeln. Sie entwickeln siRNAs, die auf Huntingtin-Botschaftsmoleküle abzielen und die Menge des HTT1a-Proteins reduzieren. Mehr Werkzeuge für unsere Huntington-Toolbox!
Won-Seok Lee: Einfluss von Proteinklumpen auf die somatische Instabilität
Der nächste Beitrag ist von Won-Seok Lee aus dem McCarroll-Labor an der Harvard Medical School. Kürzlich veröffentlichte das McCarroll-Labor eine Arbeit, die zeigt, dass anfällige Zellen bei Huntington eine somatische Expansion aufweisen – bei der die CAG-Zahl in bestimmten Zellen stark zunimmt.
In Huntington-Mausmodellen, die enorme CAG-Wiederholungen aufweisen, sehen wir viele AggregateAggregate Klumpen von Proteinen, die sich innerhalb von Zellen bei der Huntington-Krankheit und anderen degenerativen Erkrankungen bilden. Bei Menschen haben nur wenige Zellen diese riesigen CAG-Wiederholungen und wir sehen relativ wenige AggregateAggregate Klumpen von Proteinen, die sich innerhalb von Zellen bei der Huntington-Krankheit und anderen degenerativen Erkrankungen bilden. Wir wissen jedoch nicht, ob die Zellen mit den Aggregaten auch die mit den langen CAG-Werte sind.
Das Team von McCarroll wollte das herausfinden! Sie sortierten post mortem menschliche Gehirnproben, um die Zellen mit den Aggregaten zu finden. Sie fanden heraus, dass es sich bei den Gehirnzellen mit den Aggregaten um stachelige Projektionsneuronen handelt – die Zellen, die am stärksten von der Huntington-Krankheit betroffen sind – und dass diese Zellen auch sehr lange CAG-Werten haben.
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Nächstes untersuchten sie, wie Gene in diesen Zellen ein- und ausgeschaltet wurden, und stellten fest, dass diese Zellen eine ziemlich schräge genetische Signatur aufwiesen, wobei Gene eingeschaltet waren, die eigentlich ausgeschaltet sein sollten, und umgekehrt. Dies ist eine große Neuigkeit, denn es stellt einen Zusammenhang zwischen somatischer Instabilität und Aggregatbildung in menschlichen Gewebeproben UND einer gestörten Genregulation her, einem charakteristischen Merkmal der Huntington-Krankheit.
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Nächstes versuchten sie herauszufinden, welche Art von Huntingtin-ProteinHuntingtin-Protein Das Protein, das durch das Huntington-Gen hergestellt wird. in den Aggregaten zu finden war, und es sah so aus, alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. ob es sich hauptsächlich um das Fragment HTT1a handelte, von dem wir bereits von Gill gehört hatten. Zusammengenommen hilft uns diese Arbeit zu verstehen, wie jede Form von Huntingtin zur Krankheit beiträgt, was entscheidend ist, um sicherzustellen, dass wir mit Therapeutika auf die richtige Version abzielen.
„Die Botschaft, die man hier mitnehmen kann, ist, dass eine gezielte Behandlung von Msh3 in verschiedenen Huntington-Modellen dazu beitragen könnte, viele der Kennzeichen der Huntington-Krankheit wieder zu normalisieren. Diese Art von wirklich detaillierter Analyse spricht dafür, dass Msh3 ein gutes Ziel für Wissenschaftler ist, an dem sie arbeiten können, um neue Medikamente zu entwickeln. “
Spark Therapeutics: Nichtklinische Prüfung von SPK-10001
Der letzte Vortrag dieser Sitzung stammt von Liz Ramsburg von Spark Therapeutics. Spark ist ein Gentherapieunternehmen mit einem Schwerpunkt auf der Huntington-Krankheit. Spark stellt Gentherapien her, die den Huntingtin-Spiegel senken. Die TherapieTherapie Behandlungen wird in ein harmloses Virus verpackt, das dann Zellen infizieren kann, um die Maschinerie zur Senkung des Huntingtin-Spiegels freizusetzen.
Spark fand heraus, dass das Medikament am besten durch eine direkte Injektion ins Gehirn verabreicht werden kann. Obwohl dies nach einem ziemlich beängstigenden Ansatz klingt, funktionierte das Medikament in den getesteten Tiermodellen im Allgemeinen gut, wenn es auf diese Weise verabreicht wurde. Spark arbeitet an der Verbesserung seines Operationsverfahrens, um die Nebenwirkungen zu verringern.
Das Medikament SPK-10001 scheint sich gut im Gehirn zu verbreiten, und die Huntingtin-Konzentration sinkt dosisabhängig, d. h. je mehr das Medikament verabreicht wird, desto stärker sinkt die Huntingtin-Konzentration. Spark beobachtete die Tiere ein Jahr lang, nachdem sie das Medikament erhalten hatten, und es sah im Allgemeinen gut aus, was Nebenwirkungen, Veränderungen der Gehirnstrukturen usw. anbelangt. Auch die NfLNfL Biomarker für die Gesundheit des Gehirns-Werte schienen sich in einem angemessenen Zeitrahmen zu stabilisieren. Dies deutet darauf hin, dass das Medikament recht gut verträglich ist – eine gute Nachricht! Wir freuen uns darauf, mehr über die Fortschritte von Spark bei der Entwicklung von SPK-10001 zu erfahren. Liz hofft, dass sie bald in der Klinik eingesetzt werden können!
Somatische Instabilität & Mismatch-Reparatur
Dorothy Erie: Moleküle mit einem atomaren Plattenspieler sehen
Den Anfang macht heute Nachmittag Dorothy Erie, die Proteine untersucht, die an der Reaktion auf DNA-Schäden beteiligt sind. Ihr Laborteam verwendet eine Technologie namens Rasterkraftmikroskopie (atomic force microscopym AFM), um herauszufinden, wie Proteine zusammenkleben. AFM funktioniert ein wenig wie ein Plattenspieler, mit einer Nadel, die über die Oberfläche gleitet und die Topologie oder die Ecken und Kanten einer Probe sichtbar macht. Das AFM arbeitet auf molekularer Ebene, so dass die Nadel nicht dazu dient, die Musik einer Schallplatte abzuspielen, sondern uns Informationen über das liefert, was sich auf der Oberfläche befindet, in diesem Fall die Proteine.
Wie viele von Ihnen vielleicht wissen, ist die Reaktion auf DNA-Schäden ein heißes Thema bei der Huntington-Krankheit, da viele der genetischen Modifikatoren für Proteine zur Reparatur von DNA-Schäden kodieren. Zu verstehen, wie diese molekularen Maschinen funktionieren, könnte uns helfen, ihre Rolle bei der Huntington-Krankheit zu entschlüsseln.
Mit Hilfe des AFM können Dorothy und ihr Team all die verschiedenen Formen sehen, die diese Proteine bilden können. Sie bewegen sich sehr viel, was sie mit dem Tanzen der Macarena vergleicht!
Brinda Prasad: Modifikatoren im Visier
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. nächstes ist Brinda Prasad von der CHDI-Stiftung an der Reihe. Sie wird uns über verschiedene Ansätze zur therapeutischen Beeinflussung eines bestimmten DNA-Reparaturkomplexes, MutSBeta genannt, berichten. Dies ist ein heißes Zielmedikament bei der Huntington-Krankheit, das von vielen Forschern in der Wissenschaft und verschiedenen Unternehmen verfolgt wird.

MutSBeta besteht eigentlich aus zwei verschiedenen Proteinen namens MSH2 und MSH3. Sie erinnern sich vielleicht an MSH3, da dies eines der Gene ist, die alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. genetischer Modifikator der Huntington-Krankheit identifiziert wurden. Wissenschaftler glauben, dass wir den Ausbruch der Huntington-Krankheit verzögern können, wenn wir es ausschalten oder die MSH3-Konzentration verringern können.
Die Wissenschaftler haben sich viele verschiedene Möglichkeiten ausgedacht, um MutSBeta zu bekämpfen. Da MutSBeta mit anderen DNA-Reparaturmaschinen zusammenarbeitet, haben Brinda und Kollegen spezielle zirkuläre Blockiermoleküle hergestellt, die verhindern, dass es sich mit diesen Partnern verbindet.
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Nächstes untersuchten Brinda und Kollegen, wie sich die Aktivität von MutSBeta mit kleinen Molekülen ausschalten lässt. Sie haben eine ganze Reihe verschiedener Experimente durchgeführt, um diese Moleküle zu testen, um zu sehen, wie gut sie funktionieren, und um sie so zu entwickeln, dass sie die gewünschten arzneimittelähnlichen Eigenschaften haben. Dieses MutSBeta-Inhibitorprogramm läuft gut, und man hofft, einige der Leitmoleküle noch in diesem Jahr in Tiermodellen der Huntington-Krankheit testen zu können.
Ein weiteres Programm in diesem Bereich bei CHDI befasst sich mit der Herstellung von Chemikalien, die MutSBeta von der DNA abschneiden. Diese speziellen Hilfsmittel haften irreversibel an MutSBeta und hindern es an seiner normalen Aufgabe, direkt am DNA-Strang zu arbeiten, um Schäden zu reparieren.
CHDI stellt der Forschungsgemeinschaft alle Hilfsmittel und Versuchssysteme zur Verfügung, um Wissenschaftlern auf der ganzen Welt dabei zu helfen, dieses kritische Arzneimittelziel zu verfolgen und den Fortschritt zu beschleunigen. Das ist es, was wir gerne sehen wollen!
Britt Adamson: Editieren des genetischen Codes
Die nächste Teilnehmerin ist Britt Adamson von der Princeton University. Sie untersucht ebenfalls die Reaktion auf DNA-Schäden bei der Huntington-Krankheit und konzentriert sich dabei auf Mismatch-Reparaturproteine. Es wird angenommen, dass diese molekularen Maschinen für die somatische Instabilität verantwortlich sind, so dass ihre Ausschaltung nach Ansicht einiger Wissenschaftler eine Möglichkeit zur Behandlung der Huntington-Krankheit darstellt.
Ihr Labor verwendet GenomGenom Der Name, der für alle Gene vergeben wurde, die die kompletten "Bauanleitungen" einer Person oder eines Organismus enthalten-Editing-Tools, um Mismatch-Reparaturproteine zu untersuchen. Sie führen absichtlich Fehler in das GenomGenom Der Name, der für alle Gene vergeben wurde, die die kompletten "Bauanleitungen" einer Person oder eines Organismus enthalten ein und finden dann heraus, welche Proteine für die Reparatur wichtig sind und welche Arten von Änderungen sie jeweils bevorzugen. Sie testeten eine Menge verschiedener Editierungen in Zellen, denen verschiedene Mismatch-Reparaturproteine fehlten, um herauszufinden, wer was tut – sehr cool und eine großartige Ressource für das Feld!
Britt’s Team hat diese Methodik zu einer coolen neuen Plattform weiterentwickelt, um kleine Molekül-Inhibitoren von MutSBeta zu testen. Sie können schnell beurteilen, wie gut die Inhibitoren wirken und wie spezifisch sie für MutSBeta im Vergleich zu anderen Mismatch-Reparaturproteinen sind. Technologieentwicklungen wie diese werden die Entdeckung von Medikamenten in diesem Bereich wirklich vorantreiben und sicherstellen, dass Wissenschaftler auf der Suche nach Medikamenten nur die allerbesten Moleküle entwickeln, die zielgerichtet und selektiv für MutSBeta sind.
X. William Yang: Genetische Modifikatoren treiben CAG-Expansion und Krankheit an
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. nächstes hören wir von X. William Yang von der UCLA. Williams Team verwendet Mausmodelle zur Untersuchung der Huntington-Krankheit. Heute geht es um seine Arbeit über, Sie haben es erraten, Mismatch-Reparaturproteine!
„CHDI stellt der Forschungsgemeinschaft alle Hilfsmittel und Versuchssysteme zur Verfügung, um Wissenschaftlern auf der ganzen Welt dabei zu helfen, dieses kritische Arzneimittelziel zu verfolgen und den Fortschritt zu beschleunigen. Das ist es, was wir gerne sehen wollen!“
William erinnert uns an die großen humangenetischen Studien, in denen neben Huntingtin auch andere Gene identifiziert wurden, die den Zeitpunkt des Auftretens der Symptome beeinflussen können. Die Mismatch-Reparatur-Gene wurden dank Ihnen, der Huntington-Gemeinschaft, identifiziert, die sich für Studien zum natürlichen Krankheitsverlauf angemeldet haben!
William und sein Team untersuchen diese Gene in verschiedenen Mausmodellen von Huntington. Sein Team ist ein weltweiter Experte auf dem Gebiet der Mausgenetik! Obwohl Mäuse nicht mit Menschen vergleichbar sind, gibt es doch einige Ähnlichkeiten in Bezug auf die Hirnregionen, die bei der Huntington-Krankheit in diesen Modellen im Vergleich zu Menschen betroffen sind. Dies ermöglicht es ihnen, Fragen über die Rolle der Mismatch-Reparatur beim Fortschreiten der Krankheit in diesen Modellen zu stellen.
Ein wichtiges Ergebnis dieser Studie ist, dass die vollständige Entfernung von MSH3 in dem verwendeten Mausmodell offenbar dazu beiträgt, viele der molekularen Signaturen der Huntington-Krankheit wiederherzustellen. Gene, die bei Huntington-Mäusen falsch ein- oder ausgeschaltet waren, wurden durch die Beseitigung von MSH3 wieder auf das normale Niveau gebracht. Dies ist eine gute Nachricht für Menschen, die an der Entwicklung von Medikamenten arbeiten, die auf MSH3 abzielen, denn es deutet darauf hin, dass viele Merkmale der Huntington-Krankheit durch diese Art von Therapeutika korrigiert werden könnten.
Angesichts seiner Rolle bei der DNA-Reparatur überrascht es nicht, dass die Entfernung von MSH3 auch dazu beitrug, die somatische Instabilität zu verringern. Auch andere Merkmale wurden korrigiert, wie z. B. die Proteinverklumpungen, die sich im Gehirn der Mäuse bilden, sowie einige der Verhaltensweisen, die mit Huntington-Mausmodellen in Verbindung gebracht werden. William erinnert uns daran, dass Mausmodelle nützliche Instrumente zur Untersuchung der Huntington-Krankheit sind, dass es sich aber um eine menschliche Krankheit handelt und dass wir die Ergebnisse auch an Menschen und an Modellen auf menschlicher Basis validieren müssen.
Anastasia Khvorova: Zwei Ziele, ein Wirkstoff
Der letzte Vortrag des Tages stammt von Anastasia Khvorova von der University of Massachusetts. Anastasias Team arbeitet an der Entwicklung von RNAi-basierten Therapien, die gleichzeitig auf Mismatch-Reparatur-Proteine UND Huntingtin-Proteine abzielen.
Anastasias Gruppe hat ihr Repertoire an RNAi-Instrumenten erweitert, um eine ganze Reihe von Mismatch-Reparatur-Proteinen in einem Mausmodell für die Huntington-Krankheit zu reduzieren. Dies hilft uns zu verstehen, welche Proteine die besten Ziele darstellen. Zu diesem Zeitpunkt haben Sie vielleicht schon erraten, welches Protein am wichtigsten ist: …. Wenn Sie auf MSH3 getippt hätten, hätten Sie richtig gelegen!
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Nächstes untersuchten sie, was mit den Konzentrationen der verschiedenen Mismatch-Reparatur-Proteine passiert, wenn man MSH3 oder andere Proteine aus ihrem Panel ausschaltet. Auf diese Weise hoffen sie herauszufinden, welche Proteine in der Zelle zusammenhängen oder aufeinander angewiesen sind.
Um dies näher zu untersuchen, untersuchte die Gruppe von Anastasia, ob MSH3, das Huntingtin-ProteinHuntingtin-Protein Das Protein, das durch das Huntington-Gen hergestellt wird. selbst oder beides gleichzeitig in einem Mausmodell der Huntington-Krankheit ausgeschaltet werden kann. Die Mäuse schienen in allen Fällen bessere Verhaltensanzeichen und Symptome der Huntington-Krankheit zu zeigen. Die Kombinationsbehandlung schien die MSH3- oder Huntingtin-Knockdown-Behandlung allein in Bezug auf die Verringerung der toxischen Proteinverklumpungen und einiger anderer molekularer Messwerte zu übertreffen.
Diese Arbeit ist noch nicht abgeschlossen, so dass wir uns darauf freuen, mehr von Anastasia und ihren Kollegen zu erfahren, sobald sie mehr Daten haben, die sie mit allen teilen können.
Das war’s für Tag 1 der 20. Jahreskonferenz über Therapeutika für die Huntington-Krankheit! Bleiben Sie dran für Tag 2!
For more information about our disclosure policy see our FAQ…


