
Die dunkle Seite der DNA-Reparatur: ein Hinweis auf neue Behandlungsmöglichkeiten?
Ein Gen namens MSH3 unterstützt die Reparatur der DNA, kann allerdings bei Huntington zu weiteren CAG-Repeats führen. Forscher gewannen neue Kenntnisse über die Kontrolle dieser Aktivität. Sind es neue Behandlungsansätze für die Huntington-Krankheit?
Ein Gen namens „MSH3“, das den Code für ein DNA-Instandhaltungs- und Reparatureiweiß in sich trägt, hat sich zu einer heißen Spur in der Huntington-Forschung entwickelt, nachdem mehrere Studien es alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. wichtigsten Verstärker der Krankheit identifiziert haben. In einer neuen Veröffentlichung bietet ein Team von der National University of Ireland in Galway Einblicke in die Kontrollmechanismen des Gens, die bei der Entwicklung neuer Medikamente hilfreich sein könnten.
MSH3 – das zweischneidige Schwert
Das MSH3-Eiweiß ist für die Korrektur genetischer Fehler zuständig. Es schützt unser Erbgut gegen kontinuierliche Angriffe, es verhindert Schädigungen der DNA. Um dies zu erreichen, rastert es die DNA ab und sucht nach Fehlern und nimmt sich dann andere Proteine zur Hilfe, um Fehler zu beheben. Mediziner sprechen von der DNA-Mismatch-Reparatur.

Die Huntington-Krankheit wird durch eine übermäßig verlängerte DNA-Sequenz verursacht, in der immer wieder die Basenfolge C-A-G wiederholt wird und die sich auf dem Huntington-Gen befindet. In den Hirnregionen, die von der Krankheit betroffen sind, verhält sich dieser DNA-Abschnitt unstabil, was dazu führt, dass im Laufe des Lebens immer mehr CAG-Wiederholungen (CAG-Repeats) hinzu gefügt werden. Man spricht hier von somatischer Instabilität. Bei den meisten Erkrankungen, die auf einer erhöhten Repeatzahl beruhen, gilt, dass sie schwerwiegender werden, je höher die Anzahl wird.
Wenn MSH3 in unseren Zellen bei den CAG-Repeats ankommt, detektiert es einen Fehler in der DNA und versucht den Fehler zu beheben. Leider scheitert MSH3 an dieser Aufgabe und fügt stattdessen eher weitere CAG-Wiederholungen hinzu, sodass alles noch schlimmer wird!
Wie kann man das verhindern?
Wenn man ein Medikament entwickeln könnte, dass MSH3 davon abhält, diesen Fehler zu machen, könnte die somatische Expansion verhindert und damit zumindest ein Fortschreiten der Huntington-Krankheit vermieden werden. Aber wie kann das gelingen, ohne die normale, nützliche Funktion von MSH3 zu stören? Normalerweise können, wenn DNA-Reparaturgene fehlen, alle möglichen Schwierigkeiten auftreten, da sich DNA-Schäden häufen, beispielsweise könnte das Krebs verursachen. Was bei MSH3 besonders ist, ist dass seine nützliche Arbeit vollständig durch andere Gene und Proteine abdeckbar scheint. Es zeigen sich keine schädlichen Nebenwirkungen, wenn man MSH3 entfernt. Dadurch scheint es das ideale Ziel für eine Medikation zu sein.
Was ist jetzt neu?
„Wenn man ein Medikament entwickeln könnte, dass MSH3 davon abhält, diesen Fehler zu machen, könnte die somatische Expansion verhindert und damit zumindest ein Fortschreiten der Huntington-Krankheit vermieden werden.“
Unsere Zellen können dafür sorgen, dass das Eiweiß des MSH3 langsamer arbeitet, indem sie eine chemische Verbindung, eine AcetylAcetyl eine chemische Kennzeichnung, die Proteinen hinzugefügt oder von ihnen entfernt werden kann-Gruppe, hinzufügen. Sie können es auch beschleunigen, indem sie die Gruppe wieder entfernen. Da ein schneller arbeitendes MSH3-Protein zu einer schnelleren DNA-Verlängerung führen würde, wollen Wissenschaftler das Entfernen der AcetylAcetyl eine chemische Kennzeichnung, die Proteinen hinzugefügt oder von ihnen entfernt werden kann-Gruppe verhindern. Aus früherer Forschung ist bekannt, dass ein Enzym namens HDAC3 für das Entfernen der Gruppe zuständig ist. Das Lahue Labor an der National University of Ireland in Galway testete nun eine Wirksubstanz, die eben dieses HDAC3 von seiner Arbeit abhält und untersuchte die Auswirkungen auf die DNA-Expansion.
Was wurde herausgefunden?
Sie fanden heraus, dass ihr Medikament die Erhöhung der CAG-Anzahl in menschlichen Zellen verhindern konnte und – das ist wesentlich – dass dadurch keine wichtigen DNA-Reparaturarbeiten beeinträchtigt wurden, sodass die Gefahr an Krebs zu erkranken nicht erhöht wird. Die Forscher nutzten das Medikament auch, um mehr darüber zu erfahren, wie HDAC3 die Aktivität von MSH3 kontrolliert. Eiweißmoleküle nutzen spezielle Signale namens „Nuclear Localisation Sequences“ (NLS), die sie wie eine Postleitzahl an Zielorte im ZellkernZellkern Ein Teil der Zelle, der die Gene enthält (DNA) (Nukleus) leiten, wo unsere DNA aufbewahrt wird. Sie beobachteten, dass eine AcetylAcetyl eine chemische Kennzeichnung, die Proteinen hinzugefügt oder von ihnen entfernt werden kann-Gruppe die Postleitzahl für MSH3 verändert und es aus dem Nukleus heraus lenkt, wo es nicht mehr mit der DNA interagieren kann.
Was bedeutet das?
Zusammengefasst legt die Studie nahe, dass ein neuer Kontrollmechanismus für die übermäßige DNA-Verlängerung identifiziert wurde, nämlich die Entfernung der AcetylAcetyl eine chemische Kennzeichnung, die Proteinen hinzugefügt oder von ihnen entfernt werden kann-Gruppe von MSH3 durch HDAC3. Das könnte eine Erklärung dafür sein, warum Patienten ein langsameres Fortschreiten der Huntington-Krankheit zeigen, wenn sie eine spezielle Version von MSH3 aufweisen, bei der die AcetylAcetyl eine chemische Kennzeichnung, die Proteinen hinzugefügt oder von ihnen entfernt werden kann-Gruppe nur schwer entfernt werden kann.
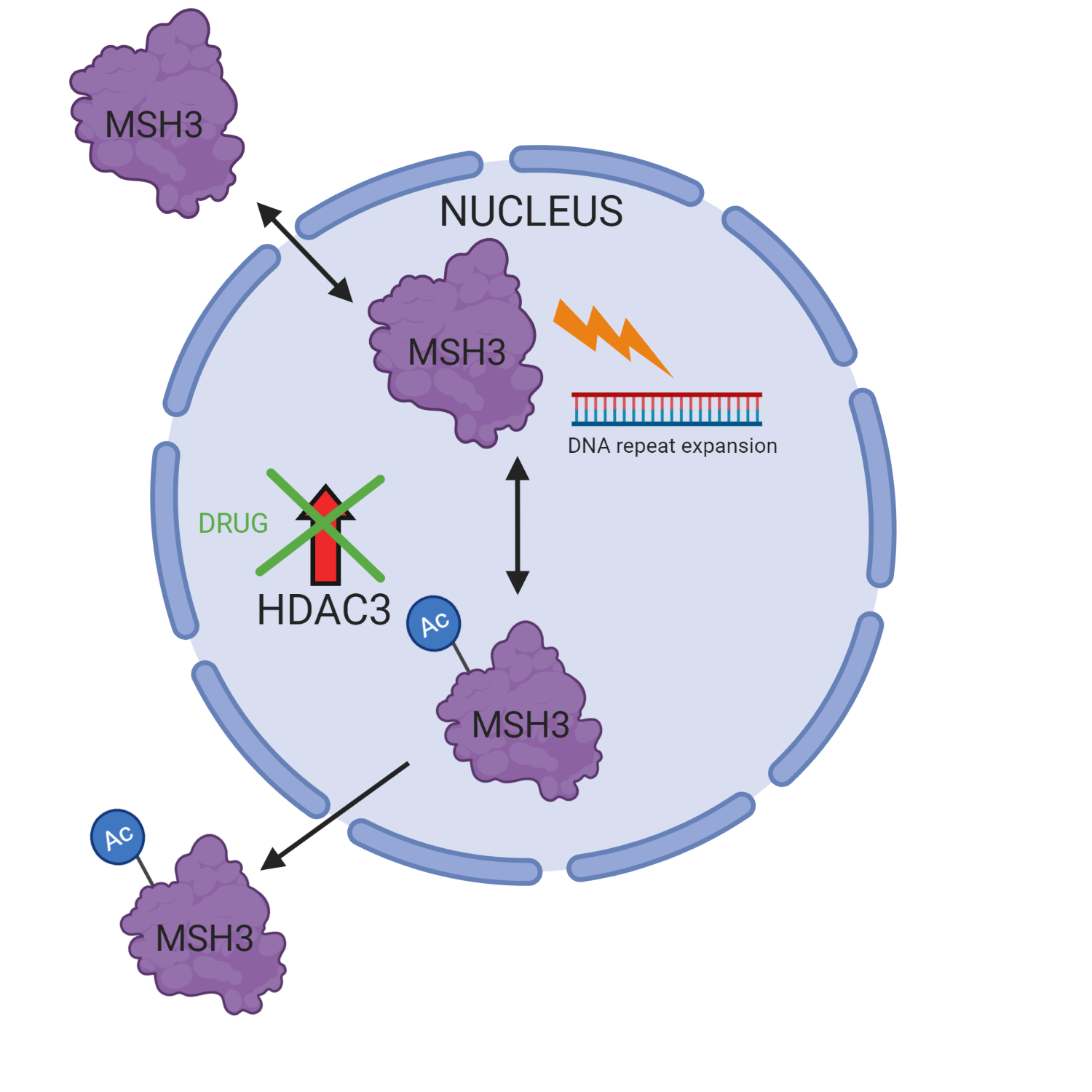
Das Verständnis von Krankheitsmechanismen auf so eine tiefgehende Art öffnet neue Türen für das Design von Medikamenten – insbesondere, da in diesem Fall eine HDAC3-Hemmung scheinbar nicht zu Beeinträchtigungen der notwendigen DNA-Mismatch-Reparatur führt. Das Medikament, das in dieser Studie untersucht wurde, hat sich alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. wirkungsvoll, selektiv und bereits recht gut erforscht. Daher könnte es bereits selbst ein vielversprechender Kandidat für klinische Studien sein.
Auch wenn das so ist, kann die Einflussnahme auf HDACHDAC Histon-Deacetylasen (HDAC) sind Enzyme, die Acetyl-Gruppen von Histonen entfernen, was verursacht, dass sie die DNA freigeben, an der sie hängen alle möglichen anderen ungewollten Nebenwirkungen auf die Zellen haben. Es muss also noch sichergestellt werden, dass das Medikament wirklich sicher ist und diese Nebeneffekte weitestgehend harmlos sind. Darüber hinaus muss eine Möglichkeit gefunden werden, die Substanz bis in die tiefen Hirnregionen vordringen zu lassen, die bei der Huntington-Krankheit betroffen sind. Dahinter verbirgt sich eine große Herausforderung.
Die Arbeit aus Galway ist also vielversprechend, es muss allerdings noch viel getan werden, bevor man sich über ein wirksames Medikament gegen Huntington freuen kann.
For more information about our disclosure policy see our FAQ…


