
Huntington's Disease Therapeutics Conference 2022 – Tag 1
Neues aus der Forschung von Tag 1 der Huntington's Disease Therapeutics Conference 2022 #HDTC2022
Guten Morgen aus dem sonnigen Palm Springs! Nach einer zweijährigen Auszeit wegen COVID, konnte die Huntington’s Disease Therapeutics Conference dieses Jahr wieder vor Ort stattfinden. Es ist die größte jährliche Versammlung von Forschern auf dem Gebiet der Huntington-Krankheit. Im Folgenden sind unsere Twitter-Beiträge aufgeführt. Auf Englisch gibt es weiterhin Updates von uns unter dem Hashtag #HDTC2022.
Tag 1 handelte von Neuigkeiten von einigen der Top-Huntington-Forschungslabore.
Huntingtin-Eiweißbausteine
Dr. Paolo Beuzer (CHDI) und Dr. Vanessa Wheeler (MGH) begannen mit der ersten Runde von Vorträgen, die sich um die Erforschung und mögliche Manipulation der CAG-Wiederholungen des mutierten Huntington-Gens drehen.
CAG-Wiederholungen – komplizierter alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. gedacht
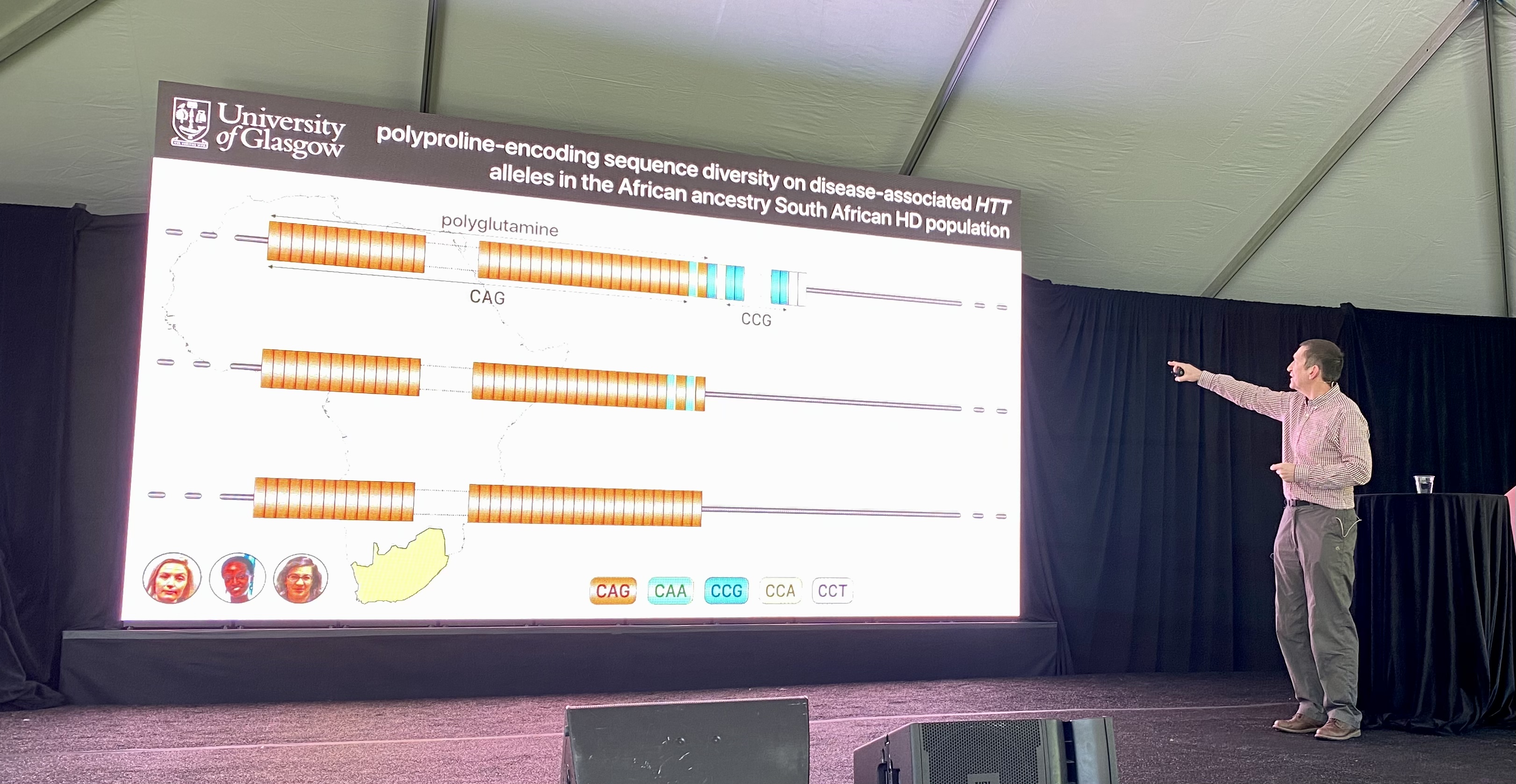
Der erste Vortrag kam von Darren Monckton von der University of Glasgow. Sein Labor untersucht die CAG-Repeats in DNA-Sequenzen, wie es sie bei der Huntington-Krankheit gibt.
Die Anzahl der Wiederholungen an sich, bestimmt noch nicht das Alter des Ausbruchs der Krankheit. Bei genauerem Hinsehen, werden die Blöcke auch nicht immer aus den Basen C-A-G gebildet, sondern manchmal aus C-A-A. Dennoch ist das Trio in beiden Fällen die Grundlage für die Bildung des Eiweißes GlutaminGlutamin Der Aminosäure-Baustein, der am Anfang des mutierten Huntingtin-Proteins zu oft wiederholt wird. Aber das Vorhandensein von C-A-A-GlutaminGlutamin Der Aminosäure-Baustein, der am Anfang des mutierten Huntingtin-Proteins zu oft wiederholt wird hat einen Einfluss auf das Ausbruchsalter. CAA tritt selten auf und sorgt bei Betroffenen für einen früheren Ausbruch der Krankheit. Es wird davon ausgegangen, dass dies durch eine Beschleunigung der somatischen Instabilität zu erklären ist, Näheres dazu hier: https://de.hdbuzz.net/291.
Die Datenbank von EnrollHD, mithilfe derer diese Erkenntnisse gewonnen wurden, besteht größtenteils aus Daten aus dem nordamerikanischen und europäischen Raum, sodass bisher nicht klar war, wie es bei Menschen aus anderen Teilen der Welt ausssieht. Die Gruppe von Monckton arbeitete nun mit einem Team in Südafrika zusammen und beobachtete in der dortigen lokalen Bevölkerung eine sehr ähnliche Seltenheit von CAA-Gruppen. Allerdings fanden sie in anderer Hinsicht Unterschiede.
Nach den CAGs enthält das Huntingtin-ProteinHuntingtin-Protein Das Protein, das durch das Huntington-Gen hergestellt wird. weitere Buchstaben, C-C-G, die einen weiteren Teil des Huntingtins namens Prolin formen. Die genaue Abfolge der Buchstaben C, C und G kann dabei variieren. Das Labor von Monckton sequenzierte die Gene von Huntington-Patienten in Südafrika und untersuchte, wie viele CCG-Wiederholungen sie hatten. Sie konnten feststellen, dass die Anzahl von der europäischer Patienten abweicht.
Mithilfe dieser Daten schauten sie sich an, inwiefern das Ausbruchsalter von der Anzahl an Prolingruppen und ihrem genauen Aufbau beeinflusst wird. Eine kleine Abweichung bei der Buchstabierung könnte demnach zu einem 10 Jahre früheren Ausbruch von Krankheitssymptomen führen. Aus diesen Erkenntnissen könnten die Diagnose und Vorhersagen zum Krankheitsverlauf verbessert werden. Der Teufel steckt beim Huntington-Gen also im Detail und es gibt mehr zu beachten alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. nur die Anzahl der CAG-Wiederholungen.
Weiterhin stellten die Forscher fest, dass der Prolin-Aufbau keinen Einfluss auf die somatische Instabilität, jedoch möglicherweise auf die Boten-RNARNA Die Chemikalie ähnlich der DNA, die die „Nachrichten“-Moleküle herstellt, die die Zellen als Arbeitskopien von Genen bei der Herstellung von Proteinen nutzen. hat. Interessanterweise schlägt dies nicht auf die Struktur des Huntingtin-Proteins selbst durch. Das bedeutet, dass nicht nur das toxische Eiweiß oder eine Veränderung dessen eine Auswirkung auf den Krankheitsverlauf hat, sondern bereits die Boten-RNARNA Die Chemikalie ähnlich der DNA, die die „Nachrichten“-Moleküle herstellt, die die Zellen als Arbeitskopien von Genen bei der Herstellung von Proteinen nutzen.. Eine veränderte Boten-RNARNA Die Chemikalie ähnlich der DNA, die die „Nachrichten“-Moleküle herstellt, die die Zellen als Arbeitskopien von Genen bei der Herstellung von Proteinen nutzen. kann aber auch schlicht zu einer anderen „Faltung“ des Eiweißes führen, von der noch niemand wirklich versteht, was sie bedeutet.
Dr. Monckton schließt damit, dass nicht alles so einfach ist, wie es scheint.
Untersuchung von Huntington in einzelnen Zellen
Steve McCarroll war der Nächste. Er arbeitet mit der Harvard Medical Schol und dem Broad Institute zusammen. Er berichtet von seiner Forschung in Bezug auf die Huntington-Krankheit in einer einzelnen Zelle.
Das Gehirn besteht aus einer Menge unterschiedlicher Zelltypen mit spezifischen Funktionen. Dr. McCarroll betont die Wichtigkeit zu verstehen, wie all diese Zelltypen durch Huntington beeinflusst werden. In seinem Labor werden spezielle Techniken verwendet, um einzelne Zelltypen zu isolieren und deren Genetik zu verstehen. Man hat sich entschieden die Arbeitsweisen öffentlich bekannt zu machen, um der gesamten Huntington-Forschung weiterzuhelfen.
Dr. McCarroll hat eine Möglichkeit gefunden, seine Analyse zu beschleunigen – er kombiniert Gehirnzellen von menschlichen Huntington-Patienten und schlüsselt die Daten nachher wieder auf. Schneller an Daten zu kommen ist ein großer Vorteil, denn somit kann die gesamte Forschung schneller voran kommen. Seine Proben entnimmt McCarroll Spendergehirnen von Huntington-Patienten, die sich vor ihrem Tod für eine Spende bereiterklärt haben.
Die Arbeitsgruppe untersucht, wie sich der Anteil unterschiedlicher Zelltypen im Gehirn mit dem Fortschreiten von Symptomen verändert. Bei den meisten Huntington-Patienten stellen sie einen bedeutenden Verlust an Zellen namens Projektionsneuronen (Englisch: medium spiny neurons (MSNs)) fest. Das Phänomen ist der Forschung bereits seit einer Weile bekannt, aber Dr. McCarroll konnte zusätzlich auch Veränderungen in anderen Zelltypen aufzeigen. Der Verlust von Zellen wird demnach durch Veränderungen begleitet, die für das an- und abschalten bestimmter Gene sorgen. Dr. McCarroll’s Gruppe hat eine ausführliche Rasterung der Genveränderungen in jedem einzelnen Zelltyp über den Verlauf der Krankheit hinweg vorgenommen – wow!
So können Gene in bestimmten Zellen identifiziert werden, die die Krankheit beeinflussen. Beispielsweise verändert sich die Anzahl der CAG-Wiederholungen auf dem Huntington-Gen, wenn ein Patient älter wird.
Bestimmte Huntington-Patienten entwickeln im Laufe der Zeit eine höhere Anzahl von CAGs, insbesondere im Gehirn (somatische Expansion). Dadurch kann das Ausbruchsalter herabgesetzt werden. Wenn es möglich wird, zu verstehen, wie diese zusätzlichen Wiederholungen zustanden kommen, können vielleicht Medikamente entwickelt werden, die das Ausbruchsalter erhöhen. Es gibt sogenannte genetische Modifizierer, die bereits bekannt sind und in McCarroll’s Projekt weiter untersucht werden.
Sie konnten bereits feststellen, dass die somatische Expansion viel stärker in Projektionsneuronen auftritt, jenem Zelltyp also, der im Verlauf der Krankheit in hohem Maße abstirbt. Weiterhin betrachtet die Arbeitsgruppe auch unterschiedliche Bereiche im Gehirn, denn je nach Hirnareal, verhalten sich auch die gleichen Zelltypen wieder unterschiedlich beispielsweise in Bezug auf die somatische Expansion. Warum ist das so? Das soll die Forschung von McCarroll herausfinden.
Huntington-Mausmodelle
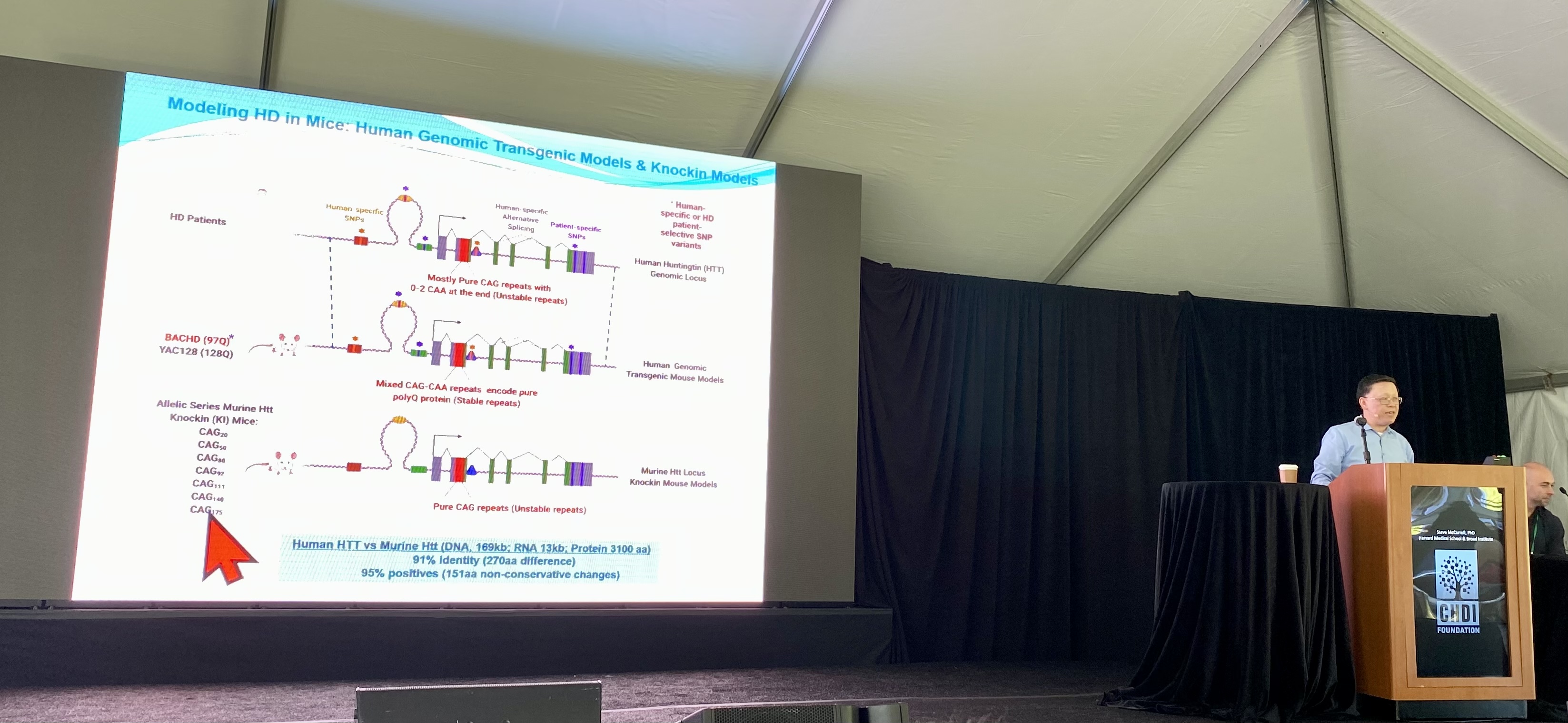
Der nächste Sprecher war Dr. William Yang von der University of California in Los Angeles, der von einem neuen Mausmodell berichtete. HDBuzz veröffentlichte bereits einen Artikel zu diesem neuen Mausmodell: https://de.hdbuzz.net/318.
Es gibt kein perfektes Mausmodell für die Huntington-Krankheit. Verschiedene Modelle können es aber ermöglichen, unterschiedliche Aspekte der Krankheit zu erforschen und ganz unterschiedliche Versuche durchzuführen. Yang’s Labor hat sich auf die Züchtung von Huntington-Mäusen spezialisiert. Das richtige Modell für das richtige Experiment zu finden ist unerlässlich, denn manche Huntington-Mäuse zeigen nur ein bestimmtes Krankheitssymptom, beispielsweise nur die Genveränderung oder nur die Anhäufung von Proteinschrott. Die wichtigste Innovation bei dem neuen Mausmodell ist, dass es somatische Expansion aufweist, dass sich die Anzahl der CAG-Wiederholungen während des Älterwerdens der Mäuse also erhöht. Man beobachtet hier auch die sich dadurch verstärkende negative Auswirkung auf das Verhalten der Mäuse und auf ihre Nervenzellen. Das ist zum ersten Mal gelungen und wurde vorher nur auf der Grundlage von menschlichen Blut- und Gewebeproben vermutet. In Yang’s labor kann nun die somatische Instabilität und deren Auswirkung auf den Bauplan des Huntingtin-Eiweißes und auf die Zellen genauer untersucht werden.
Weiterhin zeigte Yang Daten zur Gen-an- und abschaltung im Verlauf von Huntington. Damit ergänzen sie die Forschung von McCarroll und anderen Gruppen sehr gut.
Die Verarbeitung der Huntingtin-Bauanleitung
Nächster Sprecher war Dr. Gillian Bates vom Queen Square Institute of Neurology des University College London. Dr. Bates beschäftigt sich damit, welche Prozesse das Huntington-Gen in der Zelle auslöst.
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. erstes wird demnach das Gen „gespleißt“, so werden kurze Abschnitte genetischer Information zwischen dem eigentlichen Code entfernt. Dann wird der Code wieder zusammengesetzt. Dieser Prozess findet in Zellen statt, um eine gewisse Vielfalt an Anleitungen aus nur einem einzelnen Gen zu erhalten. Leider kann der Prozess bei der Huntington-Krankheit schief laufen, denn hier wird durch das SpleißenSpleißen das Zerschneiden von RNA Nachrichten, um nicht kodierende Regionen zu entfernen und kodierende Regionen zu verknüpfen. die Anleitung für ein sehr kleines Eiweiß namens „Exon 1“ erzeugt und dieses Eiweiß ist sehr schädlich für die Zellen.
Dr. Bates betrachtet die Menge an Exon 1 bei Huntington-Mausmodellen und verschiedenen Hirnarealen bei menschlichen Huntington-Patienten. Sie konnte feststellen, dass eine höhere Anzahl an CAG-Wiederholungen für eine größere Menge an Exon 1 sorgt. Bates‘ Gruppe nutzt Kombinationen bestimmter Antikörper, um die Art und Verteilung der Proteine, die durch das SpleißenSpleißen das Zerschneiden von RNA Nachrichten, um nicht kodierende Regionen zu entfernen und kodierende Regionen zu verknüpfen. erzeugt werden, zu untersuchen. Auf diese Weise haben sie herausgefunden, dass Exon 1 der Startpunkt für die unkontrollierte Anhäufung von Eiweiß ist. Diesen Vorgang genauer zu verstehen, sollte dabei helfen, Möglichkeiten zu entwickeln, die Verklumpungen zu vermeiden.
Die Arbeitsgruppe hat ein Huntington-Mausmodell gezüchtet, bei dem der Spleißvorgang verändert ist, sodass Exon 1 nicht mehr hergestellt wird. Diese Mäuse untersuchten sie hinsichtlich der Menge an schädlichen Proteinklumpen im Vergleich zu anderen Huntington-Mäusen. Tatsächlich fanden sich viel weniger Klumpen alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. bei den bekannten HD-Mausmodellen, Exon 1 scheint bei der Klumpenbildung also eine wichtige Rolle zu spielen. AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. nächstes will die Gruppe, die Auswirkungen der Klumpen auf das Verhalten der Mäuse und auf die Kommunikation zwischen ihren Nervenzellen untersuchen. Bei Menschen finden sich Verklumpungen manchmal in der Nähe des Zellkerns. Das Bates Labor konnte zeigen, dass das bei Mäusen nicht der Fall ist. Es muss also noch eine Besonderheit des menschlichen Huntingtins geben, die bisher nicht durch Mausmodelle nachgestellt werden konnte.
Wie geht die Zelle mit Huntingtin um?
Nächste Sprecherin ist Dr. Judith Frydman von der Standford University. Sie sprach darüber, wie zusätzliche CAG-Repeats das Müllindentifizierungs- und entsorgungssystem von Gehirnzellen beeinträchtigen können.
Auch wenn die Ursache von Huntington bekannt ist, kennen Forscher noch nicht wirklich die „normale“ Funktion des Eiweißes Huntingtin. Man weiß aber, dass es an einer Reihe von biologischen Prozessen beteiligt ist – es scheint vielseitig zu sein, etwa wie ein Schweizer Taschenmesser. Daher herrscht in der Forschung Uneinigkeit darüber, ob die Krankheit durch eine Störung anderer Gene oder eine Störung anderer Eiweiße ausbricht. Dr. Frydman’s Arbeit sagt, dass Huntington zumindest teilweise durch letzteres entsteht.
Ihr Labor konzentriert sich darauf, ein Verständnis darüber zu entwickeln, wie die Huntington-Boten-RNARNA Die Chemikalie ähnlich der DNA, die die „Nachrichten“-Moleküle herstellt, die die Zellen als Arbeitskopien von Genen bei der Herstellung von Proteinen nutzen. in das Protein umgewandelt wird. Dieser Prozess heißt Übersetzung. Belastungen für Zellen (beispielsweise Virusinfektionen oder Einschränkungen bei der Fähigkeit Proteinbausteine zu bauen) können den Übersetzungsprozess beeinflussen. Für die Übersetzungen sind kleine Werkzeuge namens Ribosomen notwendig. Dr. Frydman’s Arbeit zeigte, dass es bei der Herstellung des mutierten Huntingtins zu Kollisionen und Staus bei den Ribosomen kommt. AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. die Forschergruppe sich ansah, welche Gene auf der mRNA mit oder ohne Ribosomenstau verändert wurden, stellten sie fest, dass es sich um Gene handelt, die für das Aufräumen in der Zelle zuständig sind.
Ein Protein namens eIF5A taucht bei Huntington-Modellen seltener auf. eIF5A ist wichtig, um den Ribosomenstau aufzulösen. Ein Mangel sorgt also für Probleme sowohl bei der Eiweißerzeugung alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. auch bei der Entsorgung. Dr. Frydman’s Arbeit zeigt also, das Schäden sowohl auf der mRNA-Ebene alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. auch auf der Eiweißebene durch die Genmutation verursacht werden.
Die Auswirkungen von Huntingtin
Der zweite Teil der Veranstaltung wird von Dr. Balajee Somalinga (CHDI) und Dr. Ali Brivanlou (The Rockefeller University) geleitet und bezieht sich auf die Huntingtin-mRNA und das -Protein und was diese für Gesundheit bzw. Krankheit bedeuten.
Frühe Auswirkungen
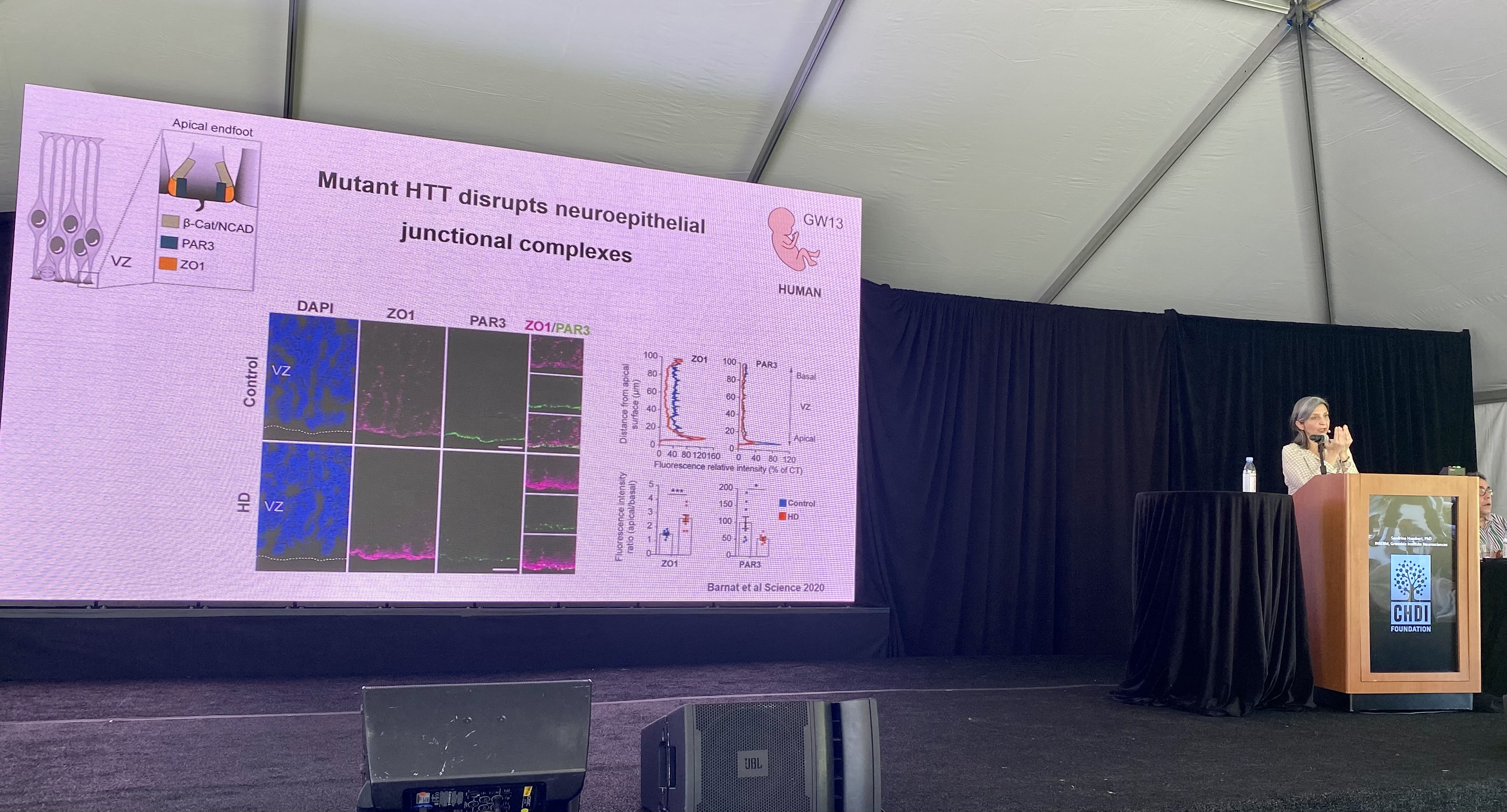
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. erste trug Sandrine Humbert von INSEMR vor und sprach über ihre Forschung an der Rolle des Huntingtins während der Entwicklung des Gehirns. Das Eiweiß hat viele Aufgaben in der Zelle, beispielsweise transportiert es verschiedene Moleküle von A nach B. Eines dieser Moleküle ist BDNFBDNF brain-derived neurotrophic factor: ein Wachstumsfaktor, der in der Lage sein könnte, die Neuronen bei der Huntington-Krankheit zu schützen, das wichtig für die Gesundheit von Nervenzellen ist. Sowohl das normale alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. auch das mutierte Huntingtin werden von den Zellen in sehr frühen Stadien des Lebens hergestellt. Humbert’s Labor denkt, dass Fehler, die in diesem Stadium auftreten bereits verantwortlich sein könnten für Symptome, die sich erst viel später im Leben zeigen. Sie entdeckten, dass Huntingtin für die Entwicklung von Nervenzellen wichtig ist, dass es außerdem ihre finale Struktur bestimmt und wie sie sich mit anderen Zellen verbinden und mit ihnen arbeiten. In ihren Huntington-Mausmodellent konnte die Gruppe zeigen, dass die Zellentwicklung durch die Mutation gestört wird, was die spätere Neurodegeneration auslösen könnte. Wir hatten bereits einen Artikel zu diesem Thema veröffentlicht: https://de.hdbuzz.net/290. Dr. Humbert’s Hypothese ist, dass die Veränderung bei den Verbindungen der Zellen in einem von Huntington betroffenen Gehirn deren Verwundbarkeit im späteren Leben verursacht.
Die neueste Arbeit aus ihrem Labor betrachtet die Wirkung von Huntingtin in gesunden und in von Huntington betroffenen Menschen. Wie beeinflusst Huntingtin das Wachstum von Nervenzellen, deren Struktur und Bewegung? Eine höhere Stabilität der Neuronenstruktur scheint sich günstig auf die spätere Zellgesundheit auszuwirken.
Zusammengefasst zeigt sich jedoch, dass sich die Nervenzellen trotz der unterschiedlichen Entwicklung im Fall von Huntington sehr resilient erweisen, sodass Krankheitssymptome erst Jahrzehnte später auftreten.
Huntingtin in anderen Spezies
Dr. Raffaele Iennaco von der Universität Milan und dem Instituto Nazionale di Genetica Molecolare sprach alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Nächster. Mithilfe von StammzellenStammzellen Zellen, die sich in verschiedene Zelltypen teilen können versucht er zu verstehen, wie die Struktur des Huntingtin Exon 1 Fragments dessen Funktionsfähigkeit beeinflusst.
Dr. Iennaco arbeitet mit dem Labor von Dr. Elena Cattaneo zusammen, wo er sich auf die Verwendung spezieller Formen von StammzellenStammzellen Zellen, die sich in verschiedene Zelltypen teilen können konzentriert, die alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. „induzierte pluripotente Stammzelleninduzierte pluripotente Stammzellen Stammzellen, die aus ausgewachsenen Zellen gezüchtet werden.“ oder iPSCs bekannt sind. Diese Zellen, die von Huntington-Patienten stammen, ermöglichen es dem Team, die Anfänge des Huntingtin-Proteins zu untersuchen. Dieses kleine Stück spiegelt nur ein winziges bisschen des Huntingtin-Proteins wider – vielleicht 3 % oder so des gesamten Proteins. Aber dieses winzige Stückchen spielt eine übergroße Rolle bei Huntingtins Aufgaben in der Zelle – insbesondere, wie es sich in der Zelle bewegt.
Um dieses kleine Stück des Huntingtin-Proteins besser zu verstehen, bestimmte das Team von Iennaco die genaue Sequenz dieser Region in 209 verschiedenen Tierarten! Dadurch erhöht sich die Zahl der Arten, für die wir diese Art von Informationen haben, dramatisch.
Die Anzahl der CAGs variiert zwischen den Arten sehr stark – bei Fischen scheinen es immer 4 CAGs zu sein, bei Eidechsen 5, während Menschen ohne HD 17-20 CAG-Wiederholungen haben. Warum verschiedene Arten unterschiedliche Mengen an CAGs benötigen, ist ein großes Rätsel, an dessen Verständnis Iennaco interessiert ist. Andere Beweise aus Iennacos Arbeit deuten darauf hin, dass das Huntingtin-Gen keine Mutationen akzeptieren kann – es gibt viel weniger Änderungen am genetischen Code von Huntingtin, alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. man zufällig erwarten würde. Ein weiterer Beweis für die Bedeutung des Huntingtin-Gens. In Weißbüschelaffen – googeln Sie das für ein sehr niedliches Affenerlebnis – gibt es tatsächlich zwei Huntingtin-Gene! Dies ist bei keiner anderen untersuchten Art der Fall, aber es deutet auf die Kraft hin, mehr alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. 200 Arten zu betrachten, um Beispiele für seltene genetische Ereignisse zu finden, um das Huntingtin-Gen besser zu verstehen. Unter Verwendung ihrer im Labor gezüchteten StammzellenStammzellen Zellen, die sich in verschiedene Zelltypen teilen können konnte das Team von Iennaco den genauen Zusammenhang zwischen der Länge der CAG-Wiederholungsregion und der Fähigkeit dieser Zellen, sich zu Gehirnzellen, sogenannten Neuronen, zu entwickeln, untersuchen. Diese Experimente helfen uns, die Bedeutung der gesamten genetischen Vielfalt zu verstehen, die in ihren Sequenzierungsstudien identifiziert wurde.
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. nächstes konzentrierten sich Iennaco und sein Team auf den Vergleich von Maus- und menschlichem Huntingtin. Seltsamerweise wurde festgestellt, dass das menschliche Huntingtin-Gen, obwohl es sich sehr ähnlich ist, giftiger ist alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. die Mausversion, aber wir wissen nicht warum. Das Team ist in der Lage, seine in Schalen gezüchteten StammzellenStammzellen Zellen, die sich in verschiedene Zelltypen teilen können dazu zu bringen, die allerersten Phasen der Gehirnentwicklung zu durchlaufen. Dies ermöglicht es ihnen, die Bedeutung kleiner Änderungen (in der CAG-Länge oder zwischen den Arten) zu untersuchen und ihre Auswirkungen auf die Gehirnentwicklung zu messen. Unter Verwendung eines sehr coolen automatisierten Systems machte das Team Bilder von etwa 5.000 verschiedenen Minigehirnen im Labor, um die Auswirkungen winziger Änderungen in der Huntingtin-Sequenz besser zu verstehen.
Viele der Aspekte des Wachstums neuer Gehirnzellen, die sie maßen, wurden stärker durch menschliches Huntingtin alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. durch Maus-Huntin beeinflusst. Dies deutet darauf hin, dass es etwas an der menschlichen Sequenz gibt, das sie auszeichnet, in ihrer Fähigkeit, für neugeborene Gehirnzellen toxisch zu sein. Das Team hat sich auf eine sehr spezifische Region des Huntingtin-Gens eingegrenzt, von der sie glauben, dass sie erklärt, warum menschliche Versionen des Huntington-Gens giftiger sind alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. die von Mäusen. Dies unterstreicht die Bedeutung genetischer Studien wie dieser bei Tieren.
Durch Huntingtin verursachte Wirkungen in Astrozyten
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. nächstes folgt ein Vortrag von Prof. Baljit Khakh von der UCLA. Sein Labor interessiert sich für einen bestimmten Typ von Stützzellen – Astrozyten genannt – bei der Huntington-Krankheit. Dies sind nicht die anfälligsten Zellen bei der Huntington-Krankheit – das sind Neuronen – aber die Aufgabe von Astrozyten im Leben ist es, Neuronen zu unterstützen. Während Astrozyten bei der Huntington-Krankheit nicht früh sterben, exprimieren sie definitiv das Huntington-Gen und zeigen eine Reihe von Veränderungen in ihrer Form und Funktion, wenn sie eine mutierte Kopie des Huntington-Gens exprimieren. Khakhs Labor möchte wissen, ob diese Veränderungen in den Astrozyten die Huntington-Krankheit beeinflussen. Khakhs Labor begann seine Arbeit, indem es sich riesige Datensätze aus den Gehirnen von Huntington-Patienten und Tiermodellen ansah, die zeigten, welche Gene an- und ausgeschaltet waren, um nach Hinweisen zu suchen, dass Astrozyten möglicherweise schlecht funktionieren. Dies schien der Fall zu sein. Es gibt Veränderungen in Astrozyten im Gehirn von Huntington-Patienten, aber spielen sie eine Rolle für das Fortschreiten der Huntington-Krankheit oder spiegeln sie nur Veränderungen in anderen Zelltypen wider?
Ein sehr cooles Huntingtin-senkendes Werkzeug namens „Zinkfinger“ kann die Expression des mutierten Huntingtin-Gens unterbinden. Wir haben bei Buzz schon früher über ZFPs geschrieben, was Sie hier nachlesen können: https://de.hdbuzz.net/275.
Das UCLA-Team war in der Lage, Viren zu entwickeln, die diese Huntingtin-senkenden Nutzlasten an verschiedene Zelltypen im Gehirn liefern, einschließlich Neuronen oder Astrozyten. Dadurch können sie das Huntingtin-Gen in verschiedenen Zelltypen senken. Diese neuen Viren reduzieren die Spiegel des Huntingtin-Gens sehr schön nur im Zielzelltyp, sodass das Team in der Lage ist, spezifische Fragen über die Beziehung zwischen der Huntingtin-Expression in bestimmten Zellen und Huntington-ähnlichen Symptomen bei Mäusen zu stellen. Das Abschalten des mutierten Huntingtins in jedem Zelltyp rettete viele der Veränderungen, die in diesem Zelltyp gefunden wurden. Wenn mutiertes Huntingtin in Neuronen abgeschaltet wird (der kranke Zelltyp bei der Huntington-Krankheit), sahen sie Verbesserungen in den Astrozyten – den Stützzellen! Das ist seltsam! Es deutet darauf hin, dass es eine Art Rückkopplungsschleife zwischen kranken Stützzellen und kranken Neuronen im Huntington-Gehirn gibt. Es zeigt auch die Macht der Manipulation bestimmter Zelltypen – die Dinge sind nicht immer so, wie wir annehmen. Das Team stellte dann die Frage – was passiert mit Huntington-ähnlichen Symptomen bei Huntington-Mäusen, wenn Huntingtin in Astrozyten oder Neuronen unter Verwendung eines ZFP gesenkt wird? Viele der von ihnen untersuchten Symptome wurden durch das Ausschalten des mutierten Huntingtin-Gens in Neuronen verbessert, aber weniger, wenn sie es in Astrozyten ausschalteten.
Das ist wichtig – Khakh liebt Astrozyten und wollte verstehen, ob sie die Huntington-Symptome antreiben. Sie führten eine sehr gute Reihe von Experimenten durch und stellten fest, dass Astrozyten verändert werden, aber dass Veränderungen in Neuronen angesichts ihrer Ergebnisse der wichtigste Faktor bleiben.
Die Huntingtin-Nachricht wird verarbeitet
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. nächstes kommt Jose Lucas vom Zentrum für Molekularbiologie Severo Ochoa (CBMSO), der darüber sprechen wird, wie die Huntingtin-Botschaft verarbeitet wird und wie sich dies bei Menschen mit der Huntington-Krankheit unterscheidet.
Der Vorgang, bei dem Genbotschaften verarbeitet werden, wird alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. SpleißenSpleißen das Zerschneiden von RNA Nachrichten, um nicht kodierende Regionen zu entfernen und kodierende Regionen zu verknüpfen. bezeichnet. Dieses Thema tauchte in einigen früheren Vorträgen auf, die sich ebenfalls mit diesem Prozess befassten, und es wird angenommen, dass das SpleißenSpleißen das Zerschneiden von RNA Nachrichten, um nicht kodierende Regionen zu entfernen und kodierende Regionen zu verknüpfen. das toxische Exon1-Fragment von Huntingtin erzeugt. Das SpleißenSpleißen das Zerschneiden von RNA Nachrichten, um nicht kodierende Regionen zu entfernen und kodierende Regionen zu verknüpfen. geht bei verschiedenen anderen Krankheiten schief, daher könnte das Verständnis der Ähnlichkeiten in diesem Prozess zwischen Krankheiten helfen, Fragen über die Huntington-Krankheit und die Symptome, die wir bei Patienten sehen, wie den Verlust von Nervenzellen, zu beantworten. Lucas und seine Kollegen untersuchten, welche Gene von Veränderungen im Spleißprozess bei der Huntington-Krankheit betroffen sind. Wenn die Botschaft eines Gens falsch gespleißt wird, bedeutet dies oft, dass weniger des vollständigen Proteinprodukts dieser Botschaft hergestellt wird. Wissenschaftler im Lucas-Labor zeigten, dass sie, wenn sie ein Gen namens RBFOX1 künstlich einschalten, die Symptome in einem Huntington-Mausmodell verbessern könnten, indem sie helfen, die Spleißfehler zu korrigieren. Vielleicht könnte diese Idee genutzt werden, um bei der Herstellung neuer Medikamente zur Behandlung der Huntington-Krankheit zu helfen?
Genbotschaften werden auch verarbeitet, um einen „Schwanz“ in ihrer genetischen Codesequenz zu entfernen, der aus vielen Buchstaben A besteht, die sich immer wieder wiederholen. Es stellt sich heraus, dass in Huntington-Modellen viele Botschaften ihre Schwänze länger behalten, alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. sie sollten, was sich darauf auswirkt, wie sie in ihre Proteinprodukte umgewandelt werden. Eines der am stärksten betroffenen Proteine, das in dieser Forschung entdeckt wurde, veranlasste die Wissenschaftler herauszufinden, dass Menschen mit der Huntington-Krankheit weniger von einem Vitamin namens Thiamin haben. Sie bestätigten dies, indem sie die Thiaminspiegel in der Rückenmarksflüssigkeit maßen, die reduzierte Werte zeigten. Die Wissenschaftler verfolgen nun in der Klinik die Antworten auf zwei unterschiedliche Fragen: Könnte der Thiaminspiegel alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. BiomarkerBiomarker Irgendeine Art von Test – inklusive Bluttest, Gedächtnistest und Gehirnscan – der das Fortschreiten einer Krankheit wie der Huntington-Krankheit messen oder vorhersagen kann. Biomarker können klinische Studien von neuen Medikamenten schneller und verlässlicher machen. für das Fortschreiten der Huntington-Krankheit verwendet werden? Und kann eine Behandlung mit Thiamin die Symptome bei Menschen mit der Huntington-Krankheit verbessern?
Obwohl dies allgemein verfügbare Vitamine sind, die die Lucas-Gruppe untersucht, sind streng kontrollierte klinische Studien erforderlich, um schlüssige Antworten zu erhalten. Hoffentlich haben wir bald Updates für Sie, wie diese mögliche Behandlung bei Menschen mit der Huntington-Krankheit funktionieren könnte.
Kontrollieren des Huntingtin-Proteinabbaus
Zum Abschluss der heutigen Vorträge wird Dr. Michael Rapé vom Howard Hughes Medical Institute, University of California, Berkeley, seine Arbeit darüber diskutieren, wie Huntingtin in der Zelle abgebaut wird und wie es zur Behandlung der Huntington-Krankheit verwendet werden könnte. Das Rapé-Labor sucht nach kleinen Molekülen, die verwendet werden können, um auf das Huntingtin-ProteinHuntingtin-Protein Das Protein, das durch das Huntington-Gen hergestellt wird. abzuzielen, damit die Zellmaschinerie es abbaut und entfernt, ein Prozess, der alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Proteinabbau bekannt ist.
Es gibt bestimmte Proteine in der Zelle, die andere Proteine für den Abbau markieren. Wenn Sie also diesen Prozess kontrollieren können, könnten Sie kontrollieren, welche Proteine die Zelle abbaut. Das wäre großartig für eine Krankheit wie die Huntington-Krankheit, bei der wir ein schädliches Protein reduzieren oder loswerden wollen! Eine Herausforderung bei Therapeutika für Gehirnerkrankungen besteht darin, die Blut-Hirn-SchrankeBlut-Hirn-Schranke Eine natürliche Barriere, gebildet durch die Verstärkung von Blutgefäßen, die den Eintritt vieler Stoffe aus dem Blut in das Gehirn verhindert. zu überwinden – die selektive Barriere, die das Gehirn vor schädlichen Dingen im Blut schützt. Die Medikamente, die das Rapé-Labor entwickelt, sind klein im Vergleich zu ASOsASOs Eine Art von Gen-Stummschaltungs-Behandlung, in der speziell entworfene DNA-Moleküle genutzt werden, um ein Gen auszuschalten (wie die von Roche und Wave entwickelten), aber immer noch groß im Vergleich zu den meisten Medikamentenmolekülen.
Glücklicherweise haben Wissenschaftler gezeigt, dass Abbauer kleiner Moleküle aus dem Blutkreislauf in das Gehirn gelangen können, was großartige Neuigkeiten für Forscher sind, die nach Abbauern suchen, um Krankheiten wie die Huntington-Krankheit zu behandeln.
Unsere Zellen stellen viele verschiedene Proteine her, sogenannte E3-Ligasen, die verwendet werden, um den Müll in der Zelle zu „markieren“ und ihn für den Abbau zu bestimmen. Wenn wir ein E3 finden könnten, das das Huntingtin-ProteinHuntingtin-Protein Das Protein, das durch das Huntington-Gen hergestellt wird. markiert, könnten wir es nutzen, um ein abbauendes Molekül zu entwickeln. Das Rapé-Labor entwickelte einen Screen, der es ihnen ermöglichen würde, E3-Ligasen zu identifizieren, die gute Ziele wären. Sie identifizierten eine E3-Ligase namens RNF126, die alle gewünschten Eigenschaften für die Entwicklung des Huntingtin-abbauenden Moleküls zu haben scheint, das RNF126 nutzbar macht. AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. nächstes testeten sie, ob RNF126 das Huntingtin-ProteinHuntingtin-Protein Das Protein, das durch das Huntington-Gen hergestellt wird. spezifisch abbauen könnte. Sie fanden heraus, dass eine erhöhte Expression von RNF126 zu einem Abbau von schädlichem Huntingtin in Zellen führte!
Aber diese Experimente wurden nur mit einem Bruchstück des schädlichen expandierten Huntingtins durchgeführt. Was passiert, wenn das gleiche Experiment mit Huntingtin-ProteinHuntingtin-Protein Das Protein, das durch das Huntington-Gen hergestellt wird. in voller Länge mit einer erweiterten CAG-WiederholungCAG-Wiederholung Der Abschnitt der DNA am Anfang des Huntington-Gens, der die Sequenz CAG viele Male wiederholt enthält und ungewöhnlich lang ist bei den Menschen, die die Huntington-Krankheit entwickeln durchgeführt wird? Die Ergebnisse repliziert! Zusammengenommen deuten diese Daten darauf hin, dass sie in der Lage waren, diese Nadel im Heuhaufen zu finden – das perfekte Enzym, das an Huntingtin bindet, um auf natürliche Weise dessen Abbau in den Zellen zu ermöglichen und eine krankheitsverursachende Proteinaggregation zu verhindern.
Die nächsten Schritte bestehen darin, RNF126 in der Arzneimittelentwicklung voranzutreiben, um zu versuchen, eine Verbindung zu identifizieren, die alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. molekularer Klebstoff bezeichnet wird und RNF126 dazu zwingt, beim Abbau des Huntingtin-Proteins zu helfen. Wir werden gespannt darauf warten, was die nächsten Schritte für dieses aufregende Molekül sind!
Bleiben Sie dran für weitere Updates!
Das ist alles für heute. Der Bericht vom 2. Tag der Konferenz folgt und dann dreht sich alles um innovative Ansätze für Huntington-Therapeutika!
For more information about our disclosure policy see our FAQ…


