
Huntington's Disease Therapeutics Conference 2021 – Tag 3
Unsere Übersicht zu Huntington's Disease Therapeutics Conference 2021 – Tag 3 #HDTC2021
Wir melden uns zurück am dritten Tag der CHDI Konferenz. Heute ging es um weitere Neuigkeiten aus klinischen Studien zur Huntington-Krankheit.
Neue Bezeichnungen für die Stadien der Huntington-Krankheit
Der erste Vortrag des Tages kam von Sarah Tabrizi und Jeff Long und handelte vom „HD Integrated Staging System“ (HD-ISS), bei dem es um eine Neudefinition der Krankheitsstadien von Huntington geht. Es soll eine weitreichendere Beschreibung des Fortschreitens ermöglichen, das durch komplexe individuelle Zusammenhänge beeinflusst werden kann.
Dem System HD-ISS gehen lange Jahre harter Arbeit durch das HD Regulatory Science Consortium voraus. Das ist ein globales Netzwerk von Forschern im akademischen und industriellen Bereich sowie von Experten der Medikamentenzulassung. Es hat sich zum Ziel gesetzt, die Rahmenbedingungen für eine reibungslose Testphase und Zulassung künftiger Medikamente abzustecken.
HD-ISS soll nun die Konzeption klinischer Studien mit Teilnehmern ermöglichen, die zwar das mutierte Huntington-Gen in sich tragen, aber noch keine Symptome der Krankheit zeigen. Solche Studien hätten zum Ziel, den Ausbruch der Krankheit zu verschieben oder ganz zu verhindern. Tabrizi erklärte, dass es sich um ein Werkzeug für die Forschung handelt, das Kriterien für die Auswahl und die medizinische Beobachtung der Teilnehmer festlegt, um Studien vergleichbar zu machen.
Bisher wurden sichtbare Kriterien herangezogen, wie beispielsweise auffällige, unkontrollierte Bewegungen. Man konnte nur bewerten, ob Genträger dieses Symptom-Stadium bereits erreicht hatten oder nicht. Es ist allerdings schon länger bekannt, dass die Huntington-Krankheit kontinuierlich verläuft, ein derartiges Schwarz-Weiß-Denken also nicht dem aktuellen Wissenstand entspricht. Auch wenn noch keine Bewegungsauffälligkeiten wahrgenommen werden können, zeigen Blutuntersuchungen und MRT-Aufnahmen, dass bei den Genträgern bereits Veränderungen im Gehirn auftreten.
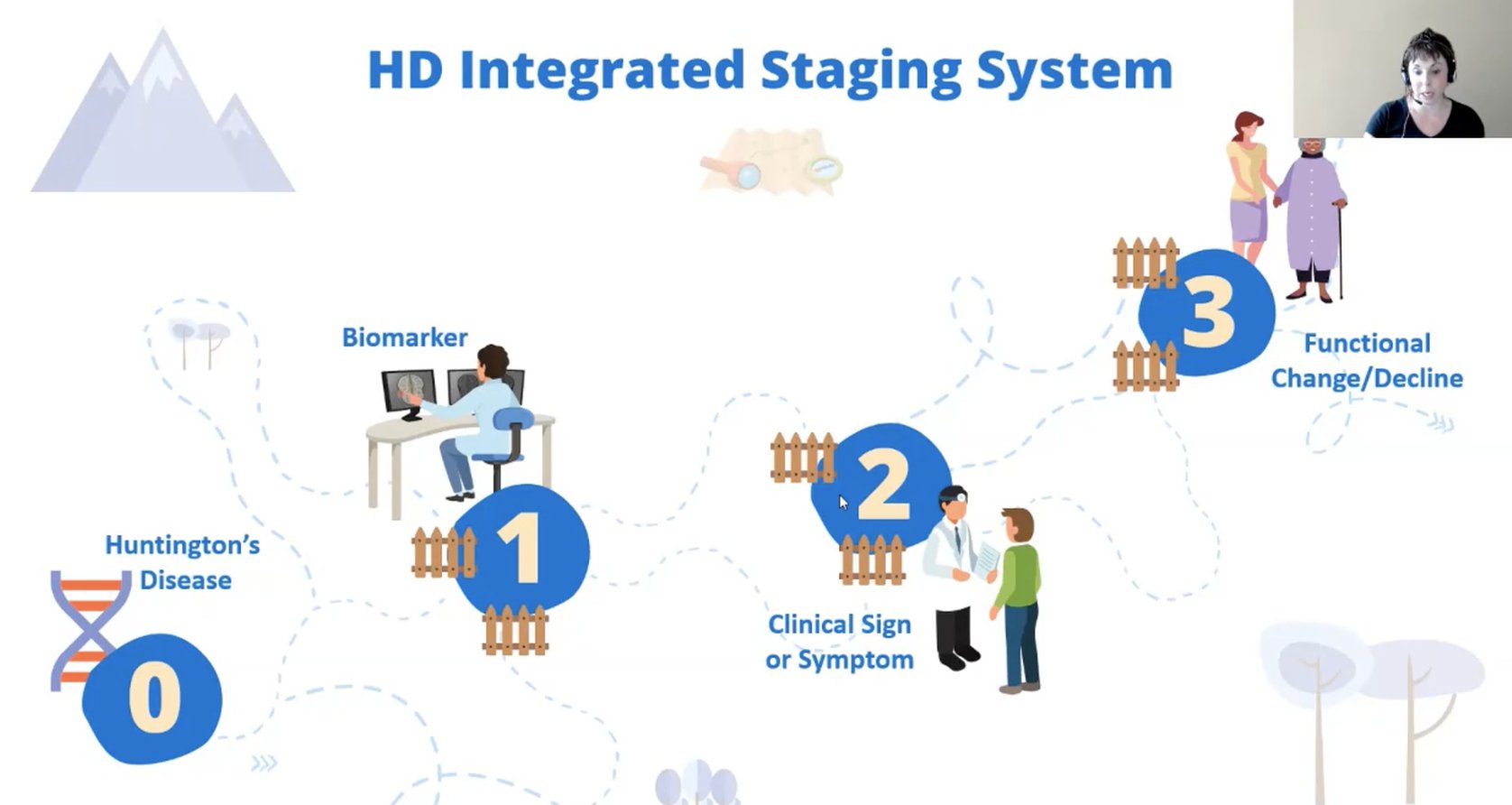
Tabrizi vertritt ein neues Konzept für die Forschung: man könnte 4 Gruppen von Genträgern unterscheiden. Als erstes gibt es die Huntington-Krankeit an sich (die lebenslangen Auswirkungen durch das mutierte Gen); bei der zweiten Gruppe ist man in der Lage biologische Effekte zu messen (Biomarker); die dritte Gruppe zeigt offensichtliche Symptome; die vierte Gruppe ist in ihren Fähigkeiten eingeschränkt.
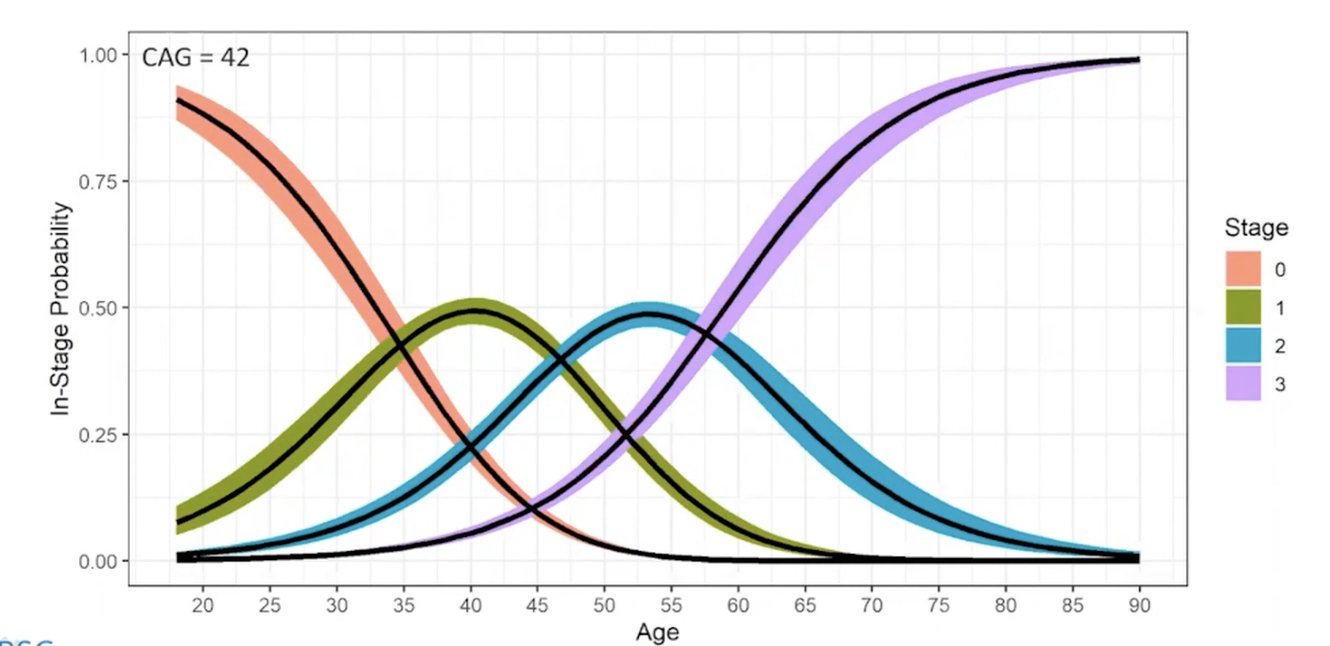
Sie benennt diese Stadien mit den Ziffern 0 bis 3, für den Übergang von einem zum nächsten Stadium werden fixe Kriterien definiert. Jeff Long erklärt hierzu, dass diese Kriterien auf Basis von großen Datenbanken systematisch festgelegt wurden. Eines wäre zum Beispiel das Volumen des Nucleus Caudatus, das über MRT ermittelt werden kann.
Daraus sollen sich einheitliche Begrifflichkeiten in der weltweiten Huntington-Forschung etablieren und ein Eingreifen im Rahmen klinischer Studien vor dem Auftreten offensichtlicher Symptome ermöglicht werden.
Neues zur Huntingtin-Verminderungsstudie HD-GeneTRX1 von uniQure
Als Nächster gab David Cooper ein paar Updates zur laufenden Studie von uniQure mit ihrem Medikament AMT-130. Wir haben kürzlich hierzu einen Artikel veröffentlicht.
Bei AMT-130 handelt es sich um die erste Gentherapie gegen die Huntington-Krankheit. Es wird in ein harmloses Virus verpackt und operativ in das Gehirn eingebracht, wo es beide Allele des Huntington-Gens stummschaltet. Cooper berichteten nochmal von erfolgreichen Studien hierzu an kleinen und großen Tiermodellen, wo sich das Medikament gut in den Gehirnen verteilte und für die Tiere verträglich war.
Cooper ging dann zu einem Lagebericht der laufenden Phase-I/II-Studie namens HD-GeneTRX1 über. Es nehmen 26 Personen teil, darunter werden einige zwar operiert, erhalten aber kein Medikament, andere erhalten eine von zwei unterschiedlichen Dosierungen des Medikamentes. Sie werden im Anschluss ein Jahr lang engmaschig beobachtet. Danach gibt es noch weitere Untersuchungen im Verlauf der folgenden 5 Jahre. Hauptziel der Studie ist die Sicherheit und Verträglichkeit des Wirkstoffes zu prüfen.
Die Teilnehmenden sind zwischen 25 und 65 Jahre alt und zeigen leichte Huntington-Symptome. Vorraussetzung waren außerdem 44 oder mehr CAG-Wiederholungen und eine bestimmte Größe gewisser Hirnareale, um die Operation sicher durchführen zu können.
Zehn Patienten wurden bisher operiert. Die Operation wird im MRT durchgeführt. Es handelt sich um brandneue Methoden und schon jetzt hat die Neurowissenschaft Wertvolles daraus gelernt. Es gibt derzeit neun Standorte in den USA, die Teilnehmer auf nehmen und uniQure will in kleinerem Umfang im Laufe des Jahres auch in Europa mit der Studie beginnen, bei der alle Teilnehmer das Medikament erhalten sollen. D. h. die Studie wird nicht verblindet, sondern offen sein.
In der Fragerunde wurde Cooper danach gefragt, ob der Abbruch der Studie von Roche kürzlich die Pläne von uniQure beeinflusst hat. Er antwortete, dass bisher alles weiter laufen soll, wie geplant. Medikament und Gabe des Medikamentes unterscheiden sich stark von Roche’s Strategie.
Blick zurück in die Daten der SIGNAL-Studie
Im Anschluss trug Maurice Zauderer von der Firma Vaccinex vor. Zauderer zeigte Ergebnisse der SIGNAL-Studie, die die Wirksamkeit des Medikaments Pepinemab gegen Huntington-Symptome untersuchte, Näheres dazu hier.
An der Studie hatten 250 Personen mit frühen Huntington-Symptomen teilgenommen. Sie bekamen Pepinemab für über ein Jahr in monatlichen Infusionen. Medizinische Kontrollen sorgten für die Beobachtung ihres Wohlbefindens und die Auswirkungen des Wirkstoffes auf die Symptome. Man konzentrierte sich insbesondere auf schwere Nebenwirkungen und mentale sowie Bewegungssymptome. Das Medikament erwies sich als sicher, aber es konnte keine konsistente Verbesserung der Symptome nachgewiesen werden.
Als Zauderer und seine Kollegen die Daten auswerteten, beobachteten sie allerdings einige leichte Verbesserungen in einer der Patientengruppen, die nur sehr leichte Symptome zeigte. Bei den Patienten mit stärkeren Symptomen zeigten sich allerdings keine Verbesserungen.
Noch mehr Zusammenarbeit in der Huntington-Forschung
Zum Abschluss der Konferenz hielt Aled Edwards vom Structural Genomics Consortium einen Vortrag über Möglichkeiten der besseren Zusammenarbeit von Forschern an Universitäten und insbesondere auch in Unternehmen in Hinblick auf die Medikamentenentwicklung und die Identifizierung möglicher Zielgene für Wirkstoffe. Oftmals hat man zu Beginn der Forschung eine riesige Anzahl von Möglichkeiten, die immer weiter heruntergebrochen wird und auf dem Weg dahin viele Erkenntnisse zur Huntington-Krankheit liefern kann.
Er plädierte für offene Wissenschaft und den freien Zugang zu Daten und dem dafür nötigen Überwinden monetärer und rechtlicher Barrieren.
Wiedersehen in 2022
Damit wurde die diesjährige Konferenz abgeschlossen und wir freuen uns bereits aufs nächste Jahr.
Weitere Informationen zu unseren Offenlegungsrichtlinien finden Sie in unseren FAQ…


