
Huntington Study Group (HSG) Conference 2024 – Tag 1
Lesen Sie aktuelle Informationen über klinische Studien und wissenschaftliche Forschung zur Huntington-Krankheit vom ersten Tag der Konferenz der Huntington Study Group 2024 #HSG2024
Die Huntington Study Group (HSG) ist ein klinisches Forschungsnetzwerk, das sich auf die Beschleunigung der Behandlung der Huntington-Krankheit konzentriert. Dieses Jahr findet die Jahreskonferenz in Cincinnati statt, wo Klinikärzte, Studien-Koordinatoren, Sozialarbeiter, Forscher und Pharmaunternehmen zusammenkommen, um aktuelle Forschungsergebnisse und Ideen auszutauschen. HDBuzz nimmt an der Konferenz teil und twittert live wissenschaftliche Updates, sobald sie stattfinden. Für diejenigen, die unsere Live-Updates nicht verfolgen konnten, haben wir unsere Tweets in diesem Artikel zusammengefasst. Lesen Sie weiter, um zu erfahren, was an Tag 1 der #HSG2024 passiert ist!
Willkommen zu HSG 2024!
Die HSG-Leitung eröffnet die Veranstaltung mit einer Reihe von Einführungen und kurzen Updates zur Tagesordnung und zur Zukunft der Organisation.

MyHDStory
In der nächsten Sitzung gab es einige kurze Updates zu den klinischen Studien, die von der HSG durchgeführt werden. Zunächst sprachen Dr. Karen Andersen aus Georgetown und Dr. Alex Dalrymple von der University of Virginia über zwei Forschungsprojekte im Rahmen der MyHDStory-Plattform.
MyHDStory ist eine Online-Studie, an der jeder aus einer Huntington-Familie in den USA zu Hause teilnehmen kann. Die Referenten erläuterten, dass die Studie es den Menschen ermöglicht, ihre Erfahrungen mit der Huntington-Krankheit mitzuteilen und an bevorstehenden Online-Studien teilzunehmen, die zur Verbesserung der Huntington-Forschung und -Pflege beitragen sollen.
KINECT-HD
Anschließend sprachen Dr. Erin Furr-Stimming von der University of Texas in Houston und Dr. Olga Klepitskaya von Neurocrine Biosciences über die KINECT-HD-Studie zu Valbenazin, das Anfang 2024 von der FDA in den USA zur Behandlung von Chorea HD zugelassen wurde
KINECT-HD hat erfolgreich gezeigt, dass Valbenazin helfen kann, unerwünschte Bewegungen zu kontrollieren. Neuere Datenanalysen zeigen, dass die stärksten Nebenwirkungen, wie z. B. Schläfrigkeitsepisoden, in den ersten Wochen der Einnahme des Medikaments auftreten und dann abklingen.
Eine Pro- und Contra-Debatte zur Huntingtin-Senkung
Für die Huntingtin-Senkung
Die nächste Sitzung ist eine Grundsatzrede mit einer Debatte über das Für und Wider von Huntingtin-senkenden Therapien. Zunächst wird Dr. Blair Leavitt, ein klinischer Huntington-Forscher an der Universität von British Columbia, über die Vorteile dieses Ansatzes sprechen.
Zur Auffrischung: Die zusätzlichen CAGs im Huntingtin-Gen führen zu einem überlangen Protein. Dr. Leavitt präsentiert erstmals Beweise aus Studien des menschlichen Gens und Proteins, die zeigen, dass diese erweiterte Form von Huntingtin schädlich sein kann. Er weist darauf hin, dass Menschen mit Huntington, die auf natürliche Weise weniger erweitertes Huntingtin-Protein produzieren, ein verzögertes Auftreten von Huntington-Symptomen aufweisen.
Er stellt auch Daten aus Studien mit verschiedenen Tier- und Zellmodellen vor, die zeigen, dass eine Senkung des Huntingtin-Spiegels sicher und oft sogar vorteilhaft ist. Die Frage, wie stark die Huntingtin-Senkung sein sollte, ist nach wie vor eine wichtige Frage, mit der sich viele Forscher im Labor und in der Klinik beschäftigen.
Blair betont auch, dass die klinischen Versuche zur Senkung des Huntingtin-Spiegels mit Tominersen (Roche), Branaplam (Novartis) und Wave-Medikamenten keine Fehlschläge waren, da die Forscher weiterhin aus ihnen lernen, indem sie die Medikamentenchemie neu gestalten, die Art und Weise, wie Therapien erprobt werden, überdenken und das Feedback der Gemeinschaft berücksichtigen.
Gegen die Huntingtin-Senkung
Als nächstes spricht Dr. Alberto Espay von der University of Cincinnati über die Herausforderungen der Huntingtin-Senkung und die Beweise, die gegen diesen Ansatz sprechen. Er erinnert uns daran, dass Huntingtin in vielen, vielen Spezies vorkommt und in verschiedenen Zellen und Organen vielfältige Funktionen hat, so dass wir vorsichtig sein müssen, wenn wir zu viel davon entfernen.
Er erklärt, dass wir sicher sind, dass das Huntingtin-Protein funktionieren kann, wenn es eine typische Länge hat, aber wir sind nicht ganz sicher, dass es nicht funktionieren kann, wenn es überlang ist. Er fordert alle auf, eine Grauzone in Betracht zu ziehen, in der ein „abnormales“ Protein sowohl gute als auch schädliche Dinge tun kann.
„Die Huntington Study Group (HSG) ist ein klinisches Forschungsnetzwerk, das sich auf die Beschleunigung der Behandlung der Huntington-Krankheit konzentriert.“
Huntingtin-Klumpen treten im Verlauf der Huntington-Krankheit in den Gehirnzellen auf, aber dies steht nicht immer in direktem Zusammenhang mit dem Verlust von Gehirnzellen. Außerdem hat Huntingtin Hunderte von „Tanzpartnern“, und nicht alle Wissenschaftler sind davon überzeugt, dass es eine gute Idee ist, in den Tanz einzugreifen und ihn zu unterbrechen!
Insgesamt argumentiert er, dass es voreilig sein könnte, überlanges Huntingtin als „toxisch“ zu bezeichnen, und dass wir vorsichtig sein sollten, wenn wir entscheiden, wann und wie viel Huntingtin gesenkt werden soll.
Er schlägt auch vor, dass wir Ansätze zur Erhöhung der Menge an „normalem“ oder „Wildtyp“-Huntingtin als Alternative zur Senkung der überlangen oder „mutierten“ Art in Betracht ziehen könnten. Das einzige Problem bei diesem Vorschlag ist, dass dies bei Mäusen, die als Modell für die Huntington-Krankheit dienten, ausprobiert wurde und keine nennenswerten Auswirkungen hatte, so dass es sich nicht lohnte, Versuche durchzuführen.
Daten bringen Therapien voran
Diese lebhafte Sitzung wird im Format einer „Debatte“ abgehalten! Dr. Leavitt räumt ein, dass Huntingtin sehr wichtig ist, kontert aber Dr. Espays Argument, dass wir versuchen könnten, das Wildtyp-Huntingtin zu vermehren, mit dem Hinweis, dass vieles darauf hindeutet, dass die erweiterte Kopie toxisch ist. Eine bloße Erhöhung des Wildtyp-Proteins wird die Toxizität der erweiterten Kopie nicht beseitigen. Es gibt jedoch wahrscheinlich ein therapeutisches Fenster für jedes Huntingtin-senkende Medikament, bei dem der richtige Zeitpunkt und das richtige Ausmaß der Senkung entscheidend sein werden.
Dr. Espay forderte alle anwesenden Unternehmen und Forscher auf, bei ihren weiteren Innovationen im Bereich der Huntingtin-senkenden Therapien an das Huntingtin-Protein und seine wichtige Rolle in den Zellen zu denken.
Der Verfechter der Huntington-Krankheit und Journalist Charles Sabine, OBE, erinnerte alle daran, dass die Huntingtin-Senkung noch Hoffnung gibt, dass aber eine Vielfalt von Forschungsansätzen uns zu einer besseren Zukunft für Menschen mit Huntington führen wird. Ein starkes Fazit dieser interessanten Debatte von Dr. Blair Leavitt: „Wir werden Therapien auf der Grundlage von Daten voranbringen!“
Arzneimittelverabreichung
In der nächsten Sitzung werden Redner über die Entdeckung von Medikamenten sprechen. Dr. Mali Jiang von der Johns Hopkins University entwickelt neuartige Verabreichungssysteme für Huntingtin-senkende und andere Ansätze zur Gentherapie. Ihre Arbeit im Labor von Dr. Lishan Lin konzentriert sich auf eine Möglichkeit, Leberzellen so umzuprogrammieren, dass sie kleine „Bläschen“, so genannte Exosomen, produzieren, die dabei helfen können, genetische Medikamente in den Blutkreislauf zu bringen.
Das Labor arbeitet an einem Medikament namens ER2001, das bereits in einer sehr kleinen Studie am Menschen in China getestet wurde. Dr. Jiang zeigte Daten über die Konzentration des Medikaments im Körper, nachdem es über einige Monate hinweg durch intravenöse Injektionen verabreicht worden war.
Ziel ist es, mit diesem neuen Ansatz Huntingtin zu senken. Das Labor von Lin und das Unternehmen, das hinter dieser Arbeit steht (ExoRNA Bioscience), hoffen, größere Studien (~30 Teilnehmer) in China und den USA durchführen zu können, wenn sie finanzielle Unterstützung finden.
PTC-518 hat einen neuen Namen! Votoplam!
Als Nächstes spricht Brian Beers von PTC therapeutics über die PIVOT-HD-Studie zu PTC-518, das jetzt Votoplam heißt. Wir haben Anfang des Jahres über die ersten positiven Ergebnisse dieser Studie geschrieben.
Brian fasst die Ergebnisse von Personen, die bis zu 12 Monate lang Votoplam einnahmen, noch einmal zusammen. Diese Menschen hatten niedrigere Huntingtin-Werte in ihrem Blut und in der Rückenmarksflüssigkeit als diejenigen, die ein Placebo (eine Pille ohne Medikament) erhielten.

An dieser Studie nahmen Teilnehmer aus einem breiten Spektrum von Huntington-Symptomen im Frühstadium teil. Die gemeldeten Nebenwirkungen, wie Kopfschmerzen, waren gering, und auf der Konferenz wurde berichtet, dass es keine Reaktionen des Immunsystems gab. Wie wir bereits im Juni berichteten, sieht PTC auch erste Trends zur Verbesserung von Körperfunktionen.
Kontrolle der CAG-Wiederholungen
Als Nächstes steht eine Forschungssitzung zum Thema Gen-Editing (Veränderung der DNA) und Bekämpfung der somatischen Instabilität (Stoppen der Expansion von CAG-Wiederholungen) an. Zunächst erläuterte Dr. Vanessa Wheeler vom Mass General Hospital und der Harvard Medical School die jüngsten Arbeiten zur Untersuchung von CAG-Expansionen auf Einzelzellebene. Diese Technik hat sich in den letzten 10 Jahren stark weiterentwickelt und liefert den Forschern Tausende von Daten. Vergleichen Sie den Blick auf die Sterne mit Ihren Augen oder mit einem leistungsstarken Teleskop. Ein gewaltiger Unterschied!
Vanessa sprach auch über GWAS-Daten – genetische Daten, bei denen jedes einzelne Gen einer Person analysiert wird. Anhand dieser großen Datensätze von vielen Menschen mit dem Gen für Huntington können Wissenschaftler feststellen, welche genetischen Informationen das Alter des Auftretens der Symptome beeinflussen.
Dadurch werden genetische Marker (so genannte Modifikatoren) identifiziert, die mit einem früheren oder späteren Alter beim Auftreten der Symptome einhergehen. Ein besseres Verständnis dieser Gene, die den Verlauf der Huntington-Krankheit beeinflussen, hilft bei der Identifizierung potenzieller Ziele für Medikamente zur Verzögerung des Auftretens von Huntington-Symptomen.
Vanessa und ihr Team untersuchen derzeit die Faktoren, die die somatische Instabilität – die ständige Ausdehnung der CAG-Wiederholung in anfälligen Gehirnzellen – beeinflussen. Sie hoffen, Gene zu identifizieren, die die somatische Instabilität kontrollieren und auf die sie therapeutisch einwirken können.
Zurzeit testen sie diese Zielgene in Mäusen, die als Modell für Huntington dienen. Wenn sie CRISPR einsetzen, um die Konzentration dieser Zielgene zu verändern, können sie das Ausmaß der somatischen Instabilität kontrollieren. Bislang haben sie 60 verschiedene Zielgene getestet. Das ist eine ganze Menge Arbeit!
Jedes Modifikatorgen, das die somatische Instabilität in Zellen oder Mäusen kontrolliert, hat das Potenzial, ein therapeutisches Ziel zu werden. Sie testen auch Kombinationen von Targets, was, wie Sie sich vorstellen können, bei 60 Targets ziemlich kompliziert wird!
Vanessa und ihr Team überlegen auch, wie einzelne oder kombinierte Targets unterschiedliche Auswirkungen auf verschiedene Zelltypen haben könnten. So kann ein Modifikator, der die somatische Instabilität in den Stützzellen des Gehirns (Glia) verlangsamt oder stoppt, eine andere Wirkung in Neuronen haben.
Sie berichtete auch über einen spezifischen Modifikator namens LIG1, der die somatische Expansion in den Gehirnen von Mäusen, die als Modell für Huntington dienen, zu reduzieren scheint. Wir warten mit Spannung auf weitere Daten zu diesem Modifikator!
Letztlich ist es Vanessas Ziel, genetische Modifikatoren der somatischen Expansion zu identifizieren, die die Instabilität kontrollieren, die Gehirnzellen länger gesund halten, das Auftreten von Symptomen verlangsamen und den von HD betroffenen Menschen mehr gesunde und glückliche Jahre bescheren.
CRISPR für die Genbearbeitung
Als nächstes ist Dr. Ricardo Mouro Pinto, ebenfalls vom Mass General Hospital und der Harvard Medical School, an der Reihe. Er wird mit uns über Gen-Editing sprechen – den Ansatz, den DNA-Bauplan zu verändern, um HD zu erforschen und schließlich zu behandeln. Aufregend!
Ricardo begann mit einer Einführung in CRISPR – einem leistungsstarken Werkzeug zur Genbearbeitung, das man sich wie eine molekulare Schere vorstellen kann. Forscher können DNA-Ziele identifizieren, die sie bearbeiten möchten, CRISPR gegen diesen DNA-Buchstabencode einsetzen und ihn durch eine neue Sequenz austauschen. Wir haben über die Fortschritte bei der Behandlung der Sichelzellenkrankheit mit CRISPR geschrieben.
„Jedes Modifikatorgen, das die somatische Instabilität in Zellen oder Mäusen kontrolliert, hat das Potenzial, ein therapeutisches Ziel zu werden.“
Ricardo geht der Frage auf den Grund, wie verschiedene DNA-Buchstaben mit unterschiedlichen Techniken ausgetauscht werden können. Seit dem Aufkommen der CRISPR-Technologie vor gerade einmal 10 Jahren hat sich in diesem Bereich viel getan.
Ricardo geht auf zwei große Herausforderungen für CRISPR in der klinischen Huntington-Forschung ein: Bereitstellung und Sicherheit. Wie bringen wir das Medikament ins Gehirn, und wie stellen wir sicher, dass das Medikament nur die gewünschten DNA-Veränderungen bewirkt?
Er weist darauf hin, dass das NIH (eine Fördereinrichtung für Wissenschaft und Medizin in den USA) eine neue Förderinitiative ins Leben gerufen hat, um die Entwicklung von Gen-Editing-Therapien zu unterstützen. Zwei der 5 derzeit finanzierten Projekte befassen sich mit Huntington!
Eines dieser Projekte konzentriert sich darauf, zusätzliche CAG-Wiederholungen aus dem Huntingtin-Gen herauszuschneiden, und das andere versucht zu verhindern, dass die CAG-Wiederholungen länger werden, um somatische Instabilitäten zu bekämpfen.
Einige Menschen mit HD haben „Unterbrechungen“ in ihren CAG-Wiederholungen, wobei stattdessen ein CAA eingefügt wird. Bei diesen Menschen treten die Symptome in der Regel später auf. Eine Gruppe von NIH-finanzierten Mitarbeitern (darunter Ricardo) arbeitet an Techniken, um CAGs in CAAs umzuwandeln.
Ricardo bezieht sich auf einige dieser „Modifikatoren“ der somatischen Instabilität, die Vanessa erwähnt hat. Er konzentriert sich auf einen dieser Modifikatoren namens MLH3 und erforscht komplexe CRISPR-Editierungstechniken, um dessen Spiegel zu senken. In Zellen kann dadurch das Wachstum von CAG-Wiederholungen verlangsamt oder gestoppt werden. Der nächste Schritt besteht darin, diese Techniken an Mäusen und an menschlichen Zellen zu testen, die die Huntington-Krankheit besser nachahmen.
Wir sind wahrscheinlich noch weit davon entfernt, diese Ansätze am Menschen zu erproben, aber es ist dennoch spannend zu sehen, dass diese Arbeit an Fahrt gewinnt und finanziert wird. Der große potenzielle Vorteil einer Gen-Editierungstherapie besteht darin, dass sie einmalig verabreicht werden könnte und eine dauerhafte Wirkung hätte.
Aktuelle Strategien für Huntington-Therapien
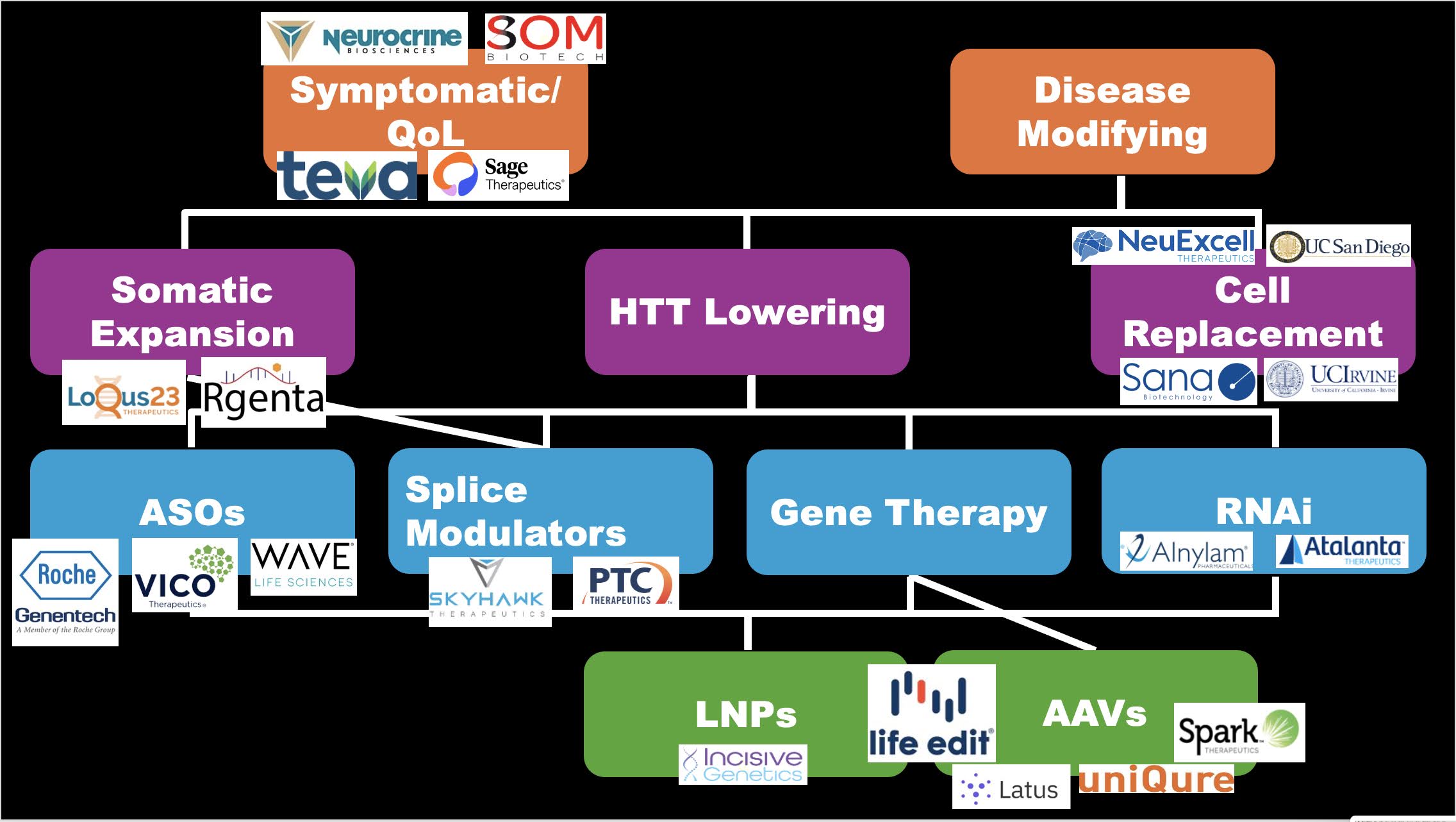
In der nächsten Sitzung werden Unternehmen, die innovative Therapien für die Huntington-Krankheit entwickeln, jeweils kurze Vorträge über ihre neuesten Entwicklungen halten. Dr. Sarah Hernandez von HDBuzz beginnt mit einem kurzen Überblick über die Grundlagen der verschiedenen Ansätze und Unternehmen in diesem Bereich.
Sarah befasst sich mit der Buchstabensuppe der Huntingtin-senkenden Techniken und Verabreichungsmethoden, wie ASOs, RNAi, AAVs und Spleißmodulatoren. Mehrere Unternehmen werden Gelegenheit haben, ausführlicher über ihre Bemühungen zur Entwicklung von Medikamenten zu sprechen. Sie erwähnt auch andere therapeutische Ansätze für die Huntington-Krankheit, wie die gezielte somatische Expansion, den Ersatz verlorener Gehirnzellen und die Verbesserung der Zell-Zell-Kommunikation.
Sarah schließt mit einer spannenden Folie, die die vielen Dutzend Unternehmen zeigt, die auf dem Gebiet der Huntington-Krankheit arbeiten. Wir leben wirklich im Zeitalter der klinischen Huntington-Studien!
Alnylam Pharmaceuticals
Der erste Redner im Forum der Innovatoren ist Dr. Kevin Sloan von Alnylam Pharmaceuticals. Sie verwenden eine Strategie namens RNAi – RNA-Interferenz -, die ein Stück genetischen Code hinzufügt, der auf die Huntingtin-Botschaft abzielt, um das Protein zu senken.
Kevin erläuterte zunächst die Einzelheiten der Funktionsweise von RNAi. Eine der größten Herausforderungen für RNAi-basierte Medikamente ist die Verabreichung. Sie müssen sicherstellen, dass das Medikament dort ankommt, wo es benötigt wird – bei Huntington ist das das ganze Gehirn. Alnylam arbeitet auch an anderen Krankheiten, und Kevin berichtet über die Daten eines RNAi-Medikaments gegen die Alzheimer-Krankheit, das in eine Phase-2-Studie geht.
Jetzt wollen sie das Gleiche für Huntington tun. Sie haben ein RNAi-Medikament für Huntingtin namens ALN-HTT02 entwickelt. Es zielt auf alle Formen von Huntingtin ab, auch auf kurze Teile, die zum Verklumpen neigen. Diese werden Huntingtin-Exon-1-Fragmente genannt und gelten als toxisch für die Gehirnzellen.
Wir haben über diese Huntingtin Exon 1 Fragmente, auch HTT1a genannt geschrieben. Dieses winzig kleine Fragment der Huntingtin-Botschaft scheint für ein Protein zu kodieren, das nur bei Menschen mit dem erweiterten Huntingtin-Gen gebildet wird.
Alnylam testet ALN-HTT02 derzeit an Affen, bevor das Medikament in klinische Studien am Menschen überführt wird. Erst diese Woche hat Alnylam bekannt gegeben, dass sie eine Phase-1-Studie für dieses Medikament beginnen werden! Diese frühe Studie wird im Vereinigten Königreich und in Kanada gestartet, die Rekrutierung in weiteren Ländern ist geplant.
Das Hauptziel ist die Sicherheit und Verträglichkeit des Medikaments, aber es wird auch untersucht, wie gut es auf Huntingtin abzielt und wie sich die Konzentrationen im Blut und im Liquor, der Flüssigkeit, die das Gehirn umspült, verändern. Sie werden klinische Tests zur Messung der Symptome durchführen, benötigen aber eine größere Studie, um zu verstehen, ob ALN-HTT02 die klinischen Merkmale der Huntington-Krankheit verändert. Es ist immer aufregend, wenn neue Studien angekündigt werden, und wir warten mit Spannung auf neue Informationen von Alnylam!
Rgenta
Als nächstes ist Dr. Travis Wager von Rgenta Therapeutics an der Reihe. Das Unternehmen entwickelt kleine Moleküle, die als Tablette eingenommen werden können, um die somatische Instabilität zu bekämpfen, d. h. die ständige Vergrößerung der CAG-Wiederholung in anfälligen Gehirnzellen.
Rgenta versucht, ein Gen namens PMS1 anzugehen – nicht das PMS, das mit Stimmungsschwankungen zu tun hat… Die Werte von PMS1 sind bei Menschen, die früher Symptome von Huntington zeigen, höher. Rgenta versucht, diese Werte zu senken, in der Hoffnung, das Auftreten von Anzeichen und Symptomen der Huntington-Krankheit zu verzögern.
Travis erklärt uns, dass es sehr wichtig ist, die richtige Stelle auf PSM1 zu wählen. Deshalb haben sie viel Zeit darauf verwendet, kleine Moleküle zu entwickeln, die an der richtigen Stelle von PMS1 angreifen. Die besten Kandidaten erfüllen auch andere Kriterien, z. B. dass sie als Pille ins Gehirn gelangen.
Rgenta hat sein Medikament in vielen verschiedenen Tiermodellen getestet. Sie haben gezeigt, dass es sehr robust ist. Wenn PMS1 um 50 % gesenkt wird, sinkt die Instabilität um 70 % – die CAG-Expansion verlangsamt sich um ein Vielfaches. Aufregend!
Travis lobte alle Forscher in der Huntington-Gemeinschaft für ihre Zusammenarbeit und die gemeinsame Nutzung ihrer Ressourcen. Er sagte, dass Rgenta ohne die Zusammenarbeit und die Bereitschaft der Huntington-Forschenden, ihre Ressourcen zu teilen, nicht da wäre, wo sie heute sind.
LifeEdit
Unser nächster Redner ist Dr. Logan Brown von LifeEdit Therapeutics, die mit Hilfe der CRISPR-Technologie Gentherapien für Huntington vorantreiben. LifeEdit arbeitet an einer CRISPR-basierten Therapie namens LETI-101, die selektiv das erweiterte Huntingtin senkt. Sie können dies tun, indem sie auf eine genetische Signatur abzielen, die sich zwischen den beiden Huntingtin-Kopien einer Person unterscheidet.
Nicht jeder weist diesen kleinen genetischen Unterschied zwischen seinen beiden Huntingtin-Kopien auf. Aus diesem Grund schätzt LifeEdit, dass LETI-101, wenn es ein Medikament werden sollte, bei 30 % der Menschen mit dem Gen für Huntington wirken würde.
Das Gute an diesem Ansatz ist, dass er aufgrund der Verwendung von CRISPR für die Genbearbeitung ein einmaliger Ansatz wäre, d. h. die Menschen bräuchten diese Behandlung theoretisch nur einmal, um Huntingtin für den Rest ihres Lebens zu senken.
Bislang wurde LETI-101 an in einer Schale gezüchteten Zellen und an Mäusen, die als Modell für Huntington dienen, getestet. Sie arbeiten aber auch an der Entwicklung einer Strategie für die Anwendung bei Menschen, was eine Gehirnoperation erfordern würde.
Sana Biotechnologie
„Wir leben wirklich im Zeitalter der klinischen Huntington-Studien!“
Als nächstes ist Dr. Joana Osorio von Sana Biotechnology an der Reihe, die an Zellersatztherapien unter Verwendung von Stammzellen arbeitet. Die Technologie von Sana entstand aus Experimenten, die zeigten, dass Stützzellen im Gehirn, die so genannten Glia, zu Krankheitsmerkmalen in Neuronen beitragen.
Mit diesem Wissen stellte Sana die Frage, ob die Transplantation von Glia-Zellen, die nicht von Huntington betroffen sind, die Krankheitsmerkmale bei Mäusen, die als Modell für Huntington dienen, verbessern könnte. Als sie dies taten, stellten sie eine Verbesserung der Bewegungen und anderer Merkmale der Krankheit fest, einschließlich einer Verlängerung der Lebensspanne der Mäuse.
Die Sana-Wissenschaftler fanden heraus, dass die Gliazellen, die nicht der Huntington-Krankheit angehören, bei der Transplantation in die Gehirne von Mäusen die Huntington-Gliazellen „verdrängten“ – sie ersetzten im Wesentlichen die kranken Huntington-Gliazellen im Gehirn! (HDBuzz berichtete bereits über diese Arbeit, als sie veröffentlicht wurde) (https://de.hdbuzz.net/347)
Sana arbeitet nun daran, diese Erkenntnisse in klinische Studien zu überführen, und obwohl sie noch nicht ganz so weit sind, haben sie einen Plan, um dieses Ziel zu erreichen. Bleiben Sie also dran!
Spark Therapeutics
Dr. Juha Savola von Spark Therapeutics ist unser nächster Redner in dieser Sitzung und berichtet über die Arbeit von Spark zur Weiterentwicklung von Gentherapien. Spark ist vor allem für die Entwicklung einer Gentherapie für eine erbliche Form des Sehverlusts bekannt (2017). Jetzt konzentrieren sie sich auf die Huntington-Krankheit.
Spark entwickelt eine Gentherapie namens SPK-10001, die das Fortschreiten von Huntington verlangsamen oder aufhalten soll, indem sie das Huntingtin reduziert. Zurzeit wird SPK-10001 an Affen getestet, und es zeigt sich, dass die HTT-Werte bis zu 12 Monate lang niedriger bleiben.
Bei den Affen werden verschiedene Dosierungen von SPK-10001 getestet und die Senkung des HTT-Spiegels in verschiedenen Hirnregionen verfolgt. Dies wird ihnen bei der Auswahl der Dosen helfen, die sie in klinischen Versuchen an Menschen testen werden. Juha teilt Einzelheiten zu den Ein- und Ausschlusskriterien für die bevorstehende Phase-I/II-Studie mit, die Spark mit Menschen mit Huntington plant.
Das Hauptziel dieser Studie wird die Sicherheit sein, aber es werden auch einige klinische Parameter untersucht, um Hinweise darauf zu erhalten, ob SPK-10001 bei der Behandlung von Huntington-Symptomen wirksam ist. Wir warten gespannt auf die Ankündigung der Rekrutierung für diese Studie!
Atalanta Therapeutics
Unsere letzte Rednerin in dieser Sitzung ist Dr. Serena Hung von Atalanta Therapeutics, die an RNAi-basierten Therapeutika gegen die Huntington-Krankheit arbeiten. Wie Sarah bereits in ihrem Überblick erläuterte, ist dies einer von mehreren Ansätzen zur Senkung des Huntingtins.
Der Vorteil des Ansatzes von Atalanta ist die Wirksamkeit. Das Unternehmen verwendet eine neuartige Technologie, mit der sich das Medikament leicht in tiefe Regionen des Gehirns ausbreiten kann. Bei der Verabreichung mittels einer Rückenmarksinjektion zeigt sich, dass das ATL-101 genannte Medikament in Tiermodellen noch bis zu sechs Monate aktiv ist.
Atalanta hat gezeigt, dass es bei Affen die HTT um 75-90 % senken kann und die NfL-Spiegel stabil bleiben. NfL ist ein Marker für die Gesundheit des Gehirns, der ansteigt, wenn Gehirnzellen geschädigt werden, und der mit fortschreitender Huntington-Krankheit zunimmt. Viele Menschen haben daher in Studien ein Auge auf NfL, denn es wäre gut, wenn dieser Wert konstant (oder sogar niedriger!) bliebe.
Atalanta plant den Beginn klinischer Studien für ATL-101 im Jahr 2025. Wir werden Sie auf dem Laufenden halten, sobald wir etwas über die Fortschritte dieser spannenden Forschung erfahren!
Schalten Sie morgen wieder ein!
Wir machen uns auf den Weg, um mehr als 80 Forschungsposter zu entdecken, die von Huntington-Wissenschaftlern und Klinikern aus der ganzen Welt präsentiert werden. In kurzen Vorträgen werden Biomarker für die Bildgebung des Gehirns und Aspekte der Enroll-HD-Plattform beleuchtet. Wir sehen uns morgen zu Sitzungen über klinische Herausforderungen bei der Huntington-Krankheit, Biomarker, Studiendesign und mehr.
Weitere Informationen zu unseren Offenlegungsrichtlinien finden Sie in unseren FAQ…


