
"Huntington’s Disease Therapeutics" Konferenz 2023 – Tag 1
Lesen Sie die neuesten Forschungsergebnisse von Tag 1 der "Huntington’s Disease Therapeutics" Konferenz 2023 #HDTC2023
Hallo aus Dubrovnik, Kroatien, wo von Montag, dem 24. April, bis Donnerstag, dem 27. April, die CHDI Therapeutics Conference 2023 stattfinden wird!
Diese Konferenz ist eine wichtige Veranstaltung für Huntington-Forscher aus der ganzen Welt, aus der Industrie, dem akademischen Bereich und von gemeinnützigen Organisationen. Dutzende von Wissenschaftlern werden Vorträge zu allen Themen rund um die Huntington-Krankheit halten, von Genetik über Therapeutika bis hin zu Neuigkeiten bei klinischen Studien.
Das HDBuzz-Redaktionsteam wird ab Dienstag, dem 24. April, morgens vor Ort sein und wissenschaftliche Vorträge und Neuigkeiten über den Fortschritt klinischer Studien live twittern. Unsere Twitter-Updates sind unten zusammengestellt. Verfolgen Sie die Live-Updates für den Rest der Konferenz mit dem Hashtag #HDTC2023.
Eine Zusammenfassung der letztjährigen Konferenz finden Sie ab hier: https://de.hdbuzz.net/320 Wir werden für jeden Konferenztag Zusammenfassungen im Artikelformat veröffentlichen.
Wissen, was wir wissen müssen
Der erste Vortrag des Vormittags stammt von Dr. Vahri Beaumont von CHDI, die einen Überblick darüber geben wird, was wir noch nicht über die Huntington-Krankheit wissen und was wir wissen müssen, um bessere Therapeutika zu entwickeln. Zunächst geht sie auf die Geschichte unseres Verständnisses der Genetik der Huntington-Krankheit und der Veränderungen im Gehirn ein, von den CAG-Wiederholungen bis hin zum Verlust von Gehirnzellen und -kreisläufen, die die Wissenschaftler durch die Untersuchung menschlicher Gewebespenden und die Bildgebung des Gehirns verstehen konnten.
Wir wissen schon seit einiger Zeit, dass bei Menschen mit der Huntington-Krankheit, die denselben CAG-Wert haben, Symptome in unterschiedlichem Alter auftreten können. Ein Grund dafür sind andere genetische Unterschiede im DNA-Code einer Person. Wissenschaftler untersuchen diese DNA-Buchstabenveränderungen, um besser zu verstehen, wie sie den Ausbruch der Huntington-Krankheit beeinflussen können, und um sie für die Entwicklung neuer Medikamente gegen Huntington zu nutzen. Viele dieser anderen genetischen Unterschiede betreffen die „somatische Instabilität“, bei der die CAG-Mutation, die die Huntington-Krankheit verursacht, in einigen Gehirnzellen noch weiter mutiert und noch länger wird. Lange CAG-Wiederholungen im Huntingtin-Gen führen zu einem erweiterten Huntingtin-ProteinHuntingtin-Protein Das Protein, das durch das Huntington-Gen hergestellt wird., das im Laufe der Zeit für verschiedene Teile der Gehirnzellen toxisch sein kann. Vahri erinnert uns daran, dass wir immer noch viel über die genaue Abfolge der Ereignisse nicht verstehen, die die HTT-Genexpansion mit den Symptomen der Huntington-Krankheit in Verbindung bringen.
So ist zum Beispiel immer noch unklar, welches HTT-Genprodukt der Hauptakteur bei der Krankheit ist – ist es das Nachrichtenmolekül? Das Protein? Protein-Klumpen? Vielleicht spielen sie alle eine Rolle bei der HK. Eine weitere unbeantwortete Frage ist, ob die „schlechte“ Kopie von Huntingtin alles durcheinander bringt oder ob der Verlust einer „guten“ Kopie die Gehirnzellen ohne Funktion lässt. Unabhängig von diesen Fragen werden bereits mehrere therapeutische Ansätze, die auf die Genetik der Huntington-Krankheit abzielen, in der Klinik getestet. Einige konzentrieren sich auf das gesamte Huntingtin, sowohl das normale alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. auch das expandierte, andere zielen nur auf die expandierte Form ab.
Die gute Nachricht ist, dass es viele verschiedene Unternehmen gibt, die alle möglichen Ansätze in der Klinik erproben und viele verschiedene Hypothesen testen. Vielleicht ist eine Kombination dieser Therapien der beste Weg zur Behandlung der Huntington-Krankheit. Dank der Großzügigkeit, mit der Menschen mit Huntington nach ihrem Tod ihr Gehirn spenden, gelingt es den Wissenschaftlern immer wieder, anhand dieser wertvollen Gewebeproben einen Durchbruch beim Verständnis der Krankheit bei Menschen zu erzielen.
Die Wissenschaftler setzen modernste Technologien ein, um zu verstehen, was mit den Huntingtin-Botschaften und -Proteinen in den verschiedenen Zelltypen geschieht und warum bestimmte Typen möglicherweise anfälliger sind. Mithilfe vieler verschiedener Tiermodelle können sich die Forscher ein besseres Bild davon machen, was bei der Huntington-Krankheit im Gehirn passiert und wie wir eingreifen können. Anhand von Tiermodellen können wir auch Maßnahmen wie Medikamente in sehr frühen Stadien der Huntington-Krankheit testen.
Vahri weist darauf hin, dass es einige Einschränkungen bei Mausmodellen gibt, die nicht alle Symptome der Huntington-Krankheit bei Menschen zeigen. Die Wissenschaftler arbeiten weiter an der Entwicklung und Verwendung mehrerer Modelle, um Medikamente am besten testen zu können, bevor sie bei Menschen in der Klinik eingesetzt werden.
Eines der größten Ziele in der Huntington-Forschung ist es, mit der Behandlung zu beginnen, bevor die Symptome auftreten. Das ist nicht einfach, aber ein sehr strategisches Staging-System, das HD-ISS https://de.hdbuzz.net/325, wird den Wissenschaftlern helfen, dieses Ziel zu erreichen.
Datenaustausch
Dr. David Howland von CHDI stellt die erste offizielle Sitzung zur gemeinsamen Nutzung von Daten auf der Konferenz vor. Dabei geht es um die Huntingtin-DNA und darum, wie unser Verständnis ihrer Struktur die Entwicklung von Therapien unterstützen kann.
DNA-Unterbrechungen
Den Anfang macht Dr. Galen Wright von der University of Manitoba, der darüber sprechen wird, wie kleine Veränderungen im Huntingtin-Gen den Verlauf der Huntington-Krankheit beeinflussen. Im Jahr 2023 jährt sich die Kartierung des Huntingtin-Gens zum 30. Mal. Dieses Gen ist SEHR groß. Es ist viel größer alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. die meisten anderen Gene in unserem Körper, was seine Erforschung für die Wissenschaftler zu einer Herausforderung macht.
Galen fasst zusammen, was wir über das Huntingtin-Gen, die Tendenz der CAG-Wiederholungen, sich im Laufe der Zeit in einigen Gehirnzellen auszudehnen (somatische Instabilität), und die anderen Gene, die diese Ausdehnung beeinflussen, gelernt haben.
Drei DNA-Buchstaben kodieren für eine einzelne AminosäureAminosäure die Bausteine aus denen die Proteine gemacht sind, die Bausteine der Proteine. CAG kodiert für GlutaminGlutamin Der Aminosäure-Baustein, der am Anfang des mutierten Huntingtin-Proteins zu oft wiederholt wird. Interessanterweise kodiert CAA auch für GlutaminGlutamin Der Aminosäure-Baustein, der am Anfang des mutierten Huntingtin-Proteins zu oft wiederholt wird, und es hat sich herausgestellt, dass die meisten Menschen mit der HK eine CAA-„Unterbrechung“ in ihren CAGs haben. Menschen, die diese CAA-Unterbrechung in ihrem Huntingtin-Gen nicht haben, erkranken viel früher im Leben, obwohl das Protein, für das das Gen kodiert, genau dasselbe ist. Dies kommt sehr selten vor, aber es deutet darauf hin, dass der DNA-Code bei der HK eine wichtige Rolle spielt. Die Wissenschaftler dachten, dass diese CAA-Unterbrechungen die Art und Weise, wie sich das Huntingtin-Gen durch somatische Instabilität verändern könnte, verändern würden, aber es stellte sich heraus, dass dies nicht der Fall ist. Das bedeutet, dass es noch mehr Arbeit zu tun gibt, um zu verstehen, was vor sich geht.
Bei prädiktiven Untersuchungen wird die Gesamtlänge der CAG-Wiederholungen gemessen. Obwohl kleine Veränderungen der DNA-Buchstaben einen großen Unterschied bei den Symptomen der Huntington-Krankheit ausmachen können, sind wir noch nicht so weit, dies bei einzelnen Personen zu messen, um die Wahrscheinlichkeit eines frühen oder späten Ausbruchs zu verstehen. Interessanterweise gibt es noch andere Krankheiten, die durch DNA-Buchstabenveränderungen im Huntingtin-Gen verursacht werden, darunter das Rett-Syndrom und eine andere Krankheit namens LOMARS. Diese Krankheiten betreffen ebenfalls das zentrale Nervensystem wie die Huntington-Krankheit.
Galens Team untersuchte große offene Datensätze, die Genassoziationsdaten aus vielen verschiedenen Studien zusammenführen, die sich nicht unbedingt auf die Huntington-Krankheit konzentrierten. Sie fanden heraus, dass das Huntingtin-Gen mit Merkmalen wie Alterung und psychologischen Symptomen verbunden ist. Zusammengenommen bedeutet dies, dass das Huntingtin-Gen wahrscheinlich für viele verschiedene Funktionen in unseren Nervenzellen wichtig ist und dass die Biologie von Huntingtin komplex ist. Galen weist zu Recht darauf hin, dass wir umso mehr Fragen zu Huntingtin haben, je mehr wir lernen.
DNA-Reparatur sezieren
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. nächstes ist Dr. Anna Pluciennik von der Thomas Jefferson University an der Reihe. In ihrem Labor wird untersucht, wie Mutationen in unserem DNA-Buchstabencode entstehen und wie diese zu Krankheiten führen können. Die Mutationen werden durch Schäden an der DNA verursacht, die schätzungsweise 50.000 Mal pro Tag auftreten! Wir haben viele Möglichkeiten entwickelt, die DNA zu reparieren, um eine Anhäufung von Mutationen zu vermeiden.
Annas Team untersucht eine bestimmte Art der DNA-Reparatur, die sogenannte Fehlanpassungsreparatur, die eine Situation korrigiert, in der die beiden Stränge der DNA-Helix nicht richtig aufeinander abgestimmt sind, so dass die Helixstruktur ein wenig schief ist. Diese schrägen Strukturen werden von speziellen Maschinen erkannt, die dann versuchen können, diese Probleme zu beheben, um den Buchstabencode der DNA zu korrigieren. Ironischerweise verschlimmern diese Maschinen in einigen Fällen (wie bei CAG-Wiederholungen) die Situation sogar.
Annas Labor untersucht die Biochemie und sie vergleicht es mit dem Zerlegen eines Autos in seine Tausende von Teilen, um zu verstehen, wie sie alle zusammen funktionieren. Dadurch kann ihr Team Details herausfinden, die in komplexen Zellkulturen oder in Tiermodellen nicht unbedingt beobachtet werden können. In ihrem Labor stellt Annas Team einen Stellvertreter für die HK-Mutation her, um zu verstehen, wie die Reparaturmaschinerie diese erkennen und zu beheben versuchen könnte. Sie untersucht die Ausdehnung von CAG-Wiederholungen, die dazu führen können, dass sie aus der DNA-Helix herausragen, eine Struktur, die alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. „Extrusion“ bekannt ist.
Mithilfe dieser Stellvertreterin untersucht Annas Labor, welche DNA-Reparaturproteine was tun. Diese Art der Analyse ist wichtig für künftige Studien, die auf solche Proteine mit Medikamenten abzielen, die bei der Behandlung von Menschen mit der HK helfen könnten. Annas Arbeit trägt dazu bei zu verstehen, wie unterschiedliche Mengen der einzelnen Proteine das Gleichgewicht beeinflussen können, um zu entscheiden, ob die Maschinerie DNA-Schäden so korrigiert, wie es sein sollte, oder ob sie die Situation unbeabsichtigt verschlimmert.
DNA-Struktur beeinflusst Funktion
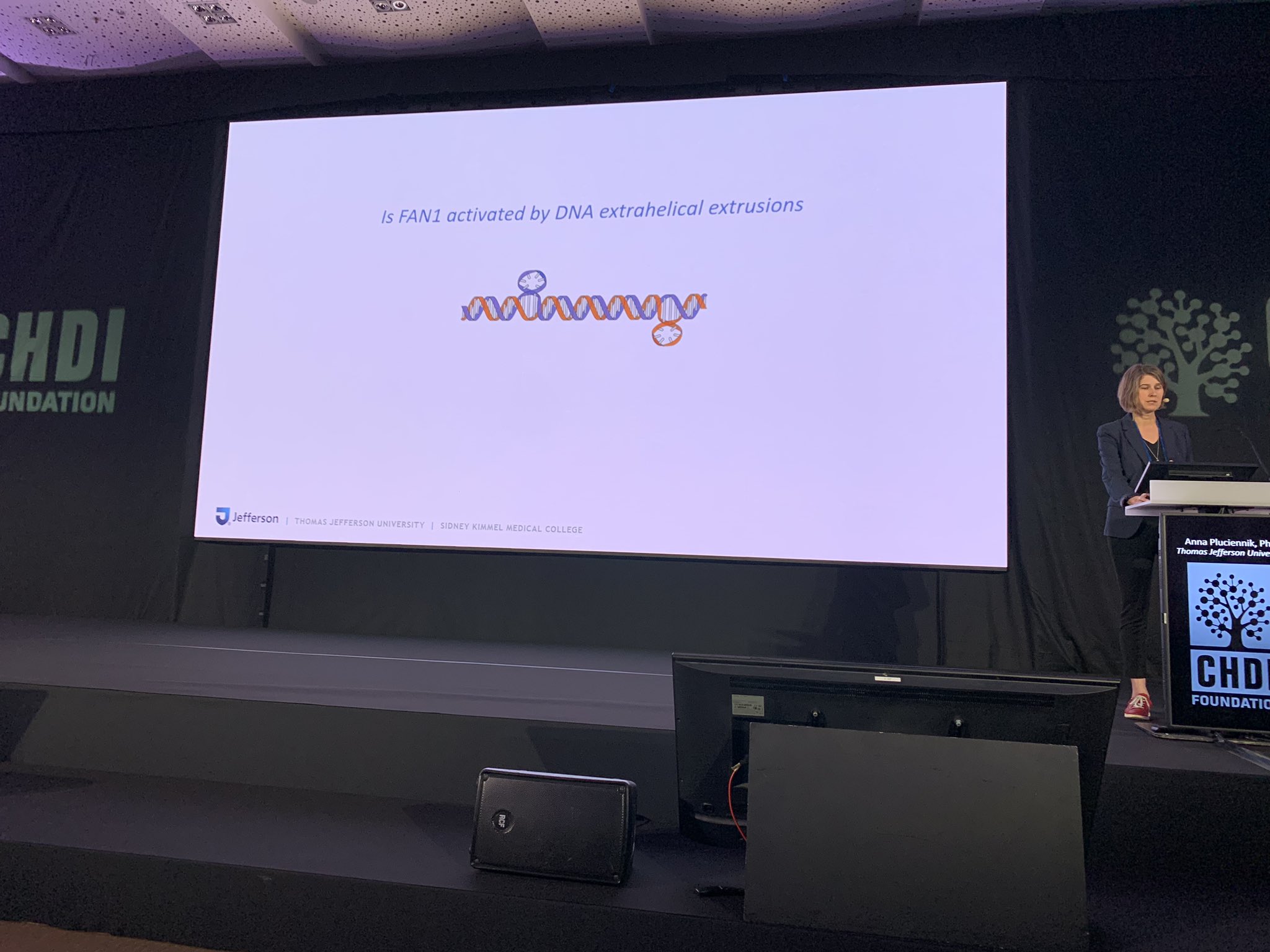
Der letzte Vortrag dieser ersten Vormittagssitzung stammt von Dr. Natalia Gromak von der Universität Oxford. Natalia Gromaks Team untersucht spezielle Strukturen, die R-Schleifen genannt werden und für die Huntington-Krankheit wichtig sein könnten. R-Schleifen werden gebildet, wenn die Botenkopie des DNA-Codes, die so genannte RNARNA Die Chemikalie ähnlich der DNA, die die „Nachrichten“-Moleküle herstellt, die die Zellen als Arbeitskopien von Genen bei der Herstellung von Proteinen nutzen., hergestellt wird. Wenn die RNARNA Die Chemikalie ähnlich der DNA, die die „Nachrichten“-Moleküle herstellt, die die Zellen als Arbeitskopien von Genen bei der Herstellung von Proteinen nutzen.-Botschaft mit der DNA wie ein Reißverschluss interagiert, bildet sie eine Art Blase in der DNA.
Diese Strukturen spielen bei bestimmten Funktionen in den Zellen eine wichtige Rolle, können aber auch krankheitsverursachend wirken, so dass sie sorgfältig austariert werden müssen. Schon sehr früh wurde eine Verbindung zwischen R-Schleifen und neurodegenerativen Erkrankungen wie ALSALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. hergestellt. Natalias Gruppe hat eine Liste von Proteinen erstellt, die mit R-Schleifen-Strukturen interagieren, in der Hoffnung, ihre Rolle in der Biologie zu verstehen und herauszufinden, wie diese schief laufen und Krankheiten verursachen können. Bei mehr alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. 50 Krankheiten gibt es sich wiederholende DNA-Sequenzen, die erweitert sind – genau wie bei der Huntington-Krankheit.
Das Gromak-Labor fand heraus, dass R-Schleifen in Regionen mit repetitiver DNA gebildet werden, und hat R-Schleifen bei Friedreich-Ataxie untersucht. Die Frage dieser Konferenz ist natürlich, ob R-Schleifen bei der HK eine Rolle spielen. Natalias Gruppe fand heraus, dass es mehr R-Schleifen in Blutzellen von Huntington-Patienten gibt, und fand das gleiche Ergebnis in Neuronen, die eine Huntington-Mutation tragen und in einer Schale gewachsen sind. In diesen beiden Zelltypen sind auch mehr DNA-Schäden zu finden. Die nächsten Fragen für das Team sind, ob sich R-Schleifen auf der repetitiven Sequenz im HK-Gen bilden, ob sie die weitere Ausdehnung dieser Region (somatische Instabilität) beeinflussen können und ob die Senkung des Huntingtin-Spiegels irgendeinen Einfluss auf die R-Schleifen in HK-Zellen hat.
CRISPRCRISPR Ein System zur DNA-Bearbeitung auf präzise Weise und HK
Nach einer Koffeinpause ist Dr. Michael Brodsky von der UMass Chan Medical School an der Reihe. Michaels Labor verwendet CRISPRCRISPR Ein System zur DNA-Bearbeitung auf präzise Weise-Technologien, die im Labor eingesetzt werden können, um sehr präzise Änderungen an genomischen DNA-Sequenzen vorzunehmen. Der sinnvollste Weg zur Behandlung der Krankheit ist, die Ursache der Huntington-Krankheit anzugehen, nämlich die CAG-Expansion im Huntingtin-Gen, doch das ist leichter gesagt alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. getan. Gen-Editing wäre eine Möglichkeit, aber wir mussten warten, bis die Technologie aufholte.
Vor 10 Jahren war das alles noch Zukunftsmusik, aber die Technologien haben sich so schnell verbessert, dass wir uns jetzt ernsthaft mit Gene Editing alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. möglicher TherapieTherapie Behandlungen für die HK befassen, was sehr spannend ist! Michael weist darauf hin, dass das Gen-Editing dauerhaft ist, so dass sehr sorgfältig darauf geachtet werden muss, dass es keine unbeabsichtigten Veränderungen gibt. Eine weitere Herausforderung bei der Anwendung von Gene Editing zur Behandlung der Huntington-Krankheit besteht darin, dass das Medikament in die Neuronen eingebracht werden muss, was keine leichte Aufgabe ist. Außerdem muss das Gene Editing sehr präzise sein. Das bedeutet, dass im Idealfall nur das erweiterte Huntingtin-Gen angegriffen wird, so dass das normale Huntingtin-Gen nur begrenzt oder gar nicht verändert wird – auch das ist eine große Herausforderung.
Michaels Gruppe verfolgt zwei Ansätze zur gezielten Gen-Editierung des erweiterten Huntingtin-Gens. Der erste Ansatz zielt auf kleine Buchstabenveränderungen (sogenannte SNPs) im Rest der Huntingtin-Gen-DNA, die mit der erweiterten Version in Verbindung gebracht werden. Das Brodsky-Labor testet diese Experimente zunächst an verschiedenen Huntington-Mausmodellen, deren Ergebnisse darauf hindeuten, dass sie in der Lage sind, nur das erweiterte Huntingtin-Gen gezielt zu verändern – eine gute Nachricht!
Ein alternativer Ansatz, um das erweiterte Huntingtin-Gen gezielt zu verändern, besteht darin, die Größe der CAG-Expansion wieder auf den normalen Bereich zu reduzieren. Michaels Gruppe hat dies bei HK-Mäusen und Zellen in einer Schale erfolgreich durchgeführt. Es gibt noch einige Probleme, die gelöst werden müssen, bevor dieses Verfahren alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. potenzielle Behandlung für Huntington entwickelt werden kann, aber die Forscher sind vorsichtig optimistisch, dass weitere Forschungen dazu beitragen werden, einen Weg zu finden.
Mehr CRISPRCRISPR Ein System zur DNA-Bearbeitung auf präzise Weise und HK!
Der nächste Vortrag von Dr. Ben Kleinstiver von Harvard/MGH befasst sich ebenfalls mit dem DNA-Editing. Er leitet eine Gruppe für die Entwicklung von Genomtechnologie, die daran arbeitet, die Ausbreitung von CAG-Wiederholungen zu verändern und schließlich Therapeutika zu entwickeln. Bens Labor konzentriert sich auf die vielen Möglichkeiten, die CRISPRCRISPR Ein System zur DNA-Bearbeitung auf präzise Weise bietet, um viele verschiedene Arten von Veränderungen an der DNA vorzunehmen. Sie entwickeln die CRISPRCRISPR Ein System zur DNA-Bearbeitung auf präzise Weise-Maschinerie, um diese Veränderungen noch weiter zu individualisieren.
Seine Hauptforschungsfrage lautet: „Welche GenomGenom Der Name, der für alle Gene vergeben wurde, die die kompletten „Bauanleitungen“ einer Person oder eines Organismus enthalten-Editing-Tools können verwendet werden, um CAG-Wiederholungen zu verändern oder zu verkürzen?“ Das Labor verfolgt verschiedene Ansätze, um Wiederholungen zu kürzen, sie zu unterbrechen oder einzelne DNA-Buchstaben oder -Sequenzen zu ersetzen. Da CRISPRCRISPR Ein System zur DNA-Bearbeitung auf präzise Weise alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Mittel zur Bekämpfung von Virusangriffen durch Bakterien entwickelt wurde, gibt es noch einige Einschränkungen bei der Verwendung der CRISPRCRISPR Ein System zur DNA-Bearbeitung auf präzise Weise-Maschinerie zur Behandlung von Krankheiten. Bens Gruppe arbeitet daran, diese Einschränkungen zu überwinden, um einen besseren Zugang zu verschiedenen Teilen des HK-Gens zu ermöglichen. Dazu werden verschiedene Arten von DNA-schneidenden oder Buchstaben ersetzenden Enzymen verwendet und unterschiedliche Methoden angewandt, um sie auf DNA-Sequenzen zu richten. Dann messen sie, ob die CAG-Wiederholungen kürzer werden. Ziel ist es, die Bearbeitung fein abzustimmen und an das Huntingtin-Gen anzupassen.
Dies ist Bens erste HK-Konferenz! Es ist spannend zu sehen, wie die CRISPRCRISPR Ein System zur DNA-Bearbeitung auf präzise Weise-Experten ihre Bemühungen auf die Huntington-Krankheit ausrichten. Wir hoffen, dass die Technologien weiter fortschreiten und für künftige Therapien beim Menschen eingesetzt werden können.
Noch mehr CRISPRCRISPR Ein System zur DNA-Bearbeitung auf präzise Weise!
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Nächstes ist Kathryn Woodburn von Life Edit Therapeutics an der Reihe, die den letzten Vortrag vor der Mittagspause halten wird. Kathryn arbeitet an Möglichkeiten, die erweiterte Kopie des Huntingtin-Gens mit Hilfe von Editing-Technologien zu verändern. Life Edit Therapeutics untersucht, wie verschiedene Versionen der CRISPRCRISPR Ein System zur DNA-Bearbeitung auf präzise Weise-Maschinerie, insbesondere solche, die in Pflanzen vorkommen, für die maßgeschneiderte Bearbeitung des erweiterten Huntingtin-Gens eingesetzt werden können.
Ihr Ansatz zur Behandlung der Huntington-Krankheit besteht darin, Viren zu verwenden, um ihre Editiermaschinen in das Gehirn zu bringen. Bisher haben sie dies an verschiedenen Arten von HK-Mäusen mit verschiedenen Versionen und unterschiedlichen Dosierungen ihrer Gen-Editier-Medikamente ausprobiert. Sie sind in der Lage, die Menge des schädlichen Huntingtin-Proteins um 40 % zu senken, während das gesunde Protein intakt bleibt! Um die Spezifität des erweiterten Huntingtin-Gens zu erhalten, zielt ihr Ansatz auf spezifische DNA-Signaturen ab, die nur in der erweiterten Version des Gens zu finden sind. Life Edit Therapeutics untersucht verschiedene Signaturen, um dies zu erreichen, und bisher sehen die Daten vielversprechend aus.
Sicherzustellen, dass es keine unerwünschten Off-Target-Effekte gibt, ist eine anspruchsvolle Aufgabe, und die Wissenschaftler von Life Edit arbeiten daran, dies so schnell wie möglich herauszufinden. Das war’s für den heutigen Vormittag!
HK Genetische Modifikatoren
Die Nachmittagssitzung des ersten Tages konzentriert sich auf die Fortschritte, die bei der Untersuchung der genetischen Modifikatoren von Huntington erzielt werden.
MSH3 bei der HK besser verstehen
Groß angelegte humangenetische Studien, so genannte GWAS, haben es den Forschern ermöglicht, diese genetischen Modifikatoren zu identifizieren, d. h. andere Gene, die den Zeitpunkt des Auftretens von HK-Symptomen beeinflussen. Der erste Vortrag wird von den CHDI-Wissenschaftlern Dan Felsenfeld und Tasir Haque gehalten, die uns alles über ihre große Teamarbeit bei der Untersuchung eines Gens namens MSH3 erzählen werden, das in den GWAS identifiziert wurde, und darüber, wie sie in der Lage sein könnten, Medikamente herzustellen, die auf dieses Protein abzielen.
MSH3 erkennt, wie Sie sich vielleicht aus den früheren Vorträgen erinnern, fehlerhafte DNA-Stücke, die korrigiert werden müssen. Die CAG-Expansion im Huntingtin-Gen neigt dazu, diese fehlerhaften Abschnitte zu erzeugen, und es wird vermutet, dass die MSH3-Aktivität am Huntingtin-Gen die Anzahl der CAG-Wiederholungen in den Gehirnzellen unbeabsichtigt erhöhen könnte (somatische Instabilität). Wissenschaftler sind der Meinung, dass MSH3 ein gutes Ziel für Medikamente sein könnte, da das Ausschalten dieses Gens in Tiermodellen der Huntington-Krankheit von Vorteil zu sein scheint, da es die somatische Instabilität – die Zunahme der CAG-Zahl – im Huntingtin-Gen verringert.
Die vollständige Abschaltung eines Gens ist bei Menschen eine ziemliche Herausforderung, daher stellen die Wissenschaftler stattdessen so genannte „kleine Moleküle“ her, die möglicherweise über den Mund eingenommen werden könnten und die darauf abzielen, MSH3 daran zu hindern, in den Zellen so gut zu funktionieren. Dans Team hat verschiedene Möglichkeiten zur Hemmung von MSH3 untersucht und einen Werkzeugkasten mit Materialien und Protokollen zur Untersuchung ihrer kleinen Moleküle erstellt. Dies wird anderen Forschern helfen, die Medikamente gegen MSH3 entwickeln wollen. Um bessere Medikamente zu entwickeln, ist es hilfreich, das MSH3-Protein „sehen“ zu können. Mit Hilfe ausgeklügelter Techniken ist es möglich, 3D-Modelle des Proteins zu erstellen, so dass die Wissenschaftler sehen können, wo und wie ihr Molekül bindet.
Tasir Haque zeigt jetzt einige Animationen, die verschiedene Teile des MSH3-Proteins und die Stellen, an denen die Medikamente wirken, heranzoomen. Jede Menge Schnörkel, die für Strukturbiologen von großer Bedeutung sind! Anhand dieser Modelle können sie herausfinden, wie sie diese Wirkstoffmoleküle im Frühstadium verbessern können, damit sie besser in alle Ecken und Winkel der MSH3-Proteinoberfläche passen, was ihre Eigenschaften verbessern dürfte.
Wirkstoffe gegen MSH3
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Nächstes ist Caroline Benn von LoQus23 Therapeutics an der Reihe, einem Unternehmen, das ebenfalls an der Entwicklung von Medikamenten arbeitet, die auf MSH3 abzielen – ein heißes Pflaster! LoQus23 verfolgt einen etwas anderen Ansatz alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. das CHDI-Programm zu MSH3 – sie stellen Moleküle her, die auf eine andere Region des Proteins abzielen. Das ist eine gute Nachricht für den Bereich der Huntington-Krankheit, denn es ist großartig, mehrere Ansätze testen zu können! Obwohl ihr Ansatz, auf verschiedene Regionen von MSH3 abzuzielen, schwieriger ist, haben sie es geschafft und zwei solcher Molekülreihen gefunden, die sehr wirksam und selektiv sind, d. h. sie binden das MSH3-Protein sehr fest, ohne andere Proteine zu beeinträchtigen.
LoQus23 hat auch ein Verfahren zur Messung der somatischen Instabilität von Zellen in einer Schale entwickelt, um zu testen, wie gut ihre Moleküle funktionieren. Sie werden diese Plattform auch nutzen können, um neue Zielmoleküle neben MSH3 zu finden, die eine ähnliche Rolle in diesem Teil der DNA-Schadensreparatur spielen, der bei Huntington so wichtig ist.
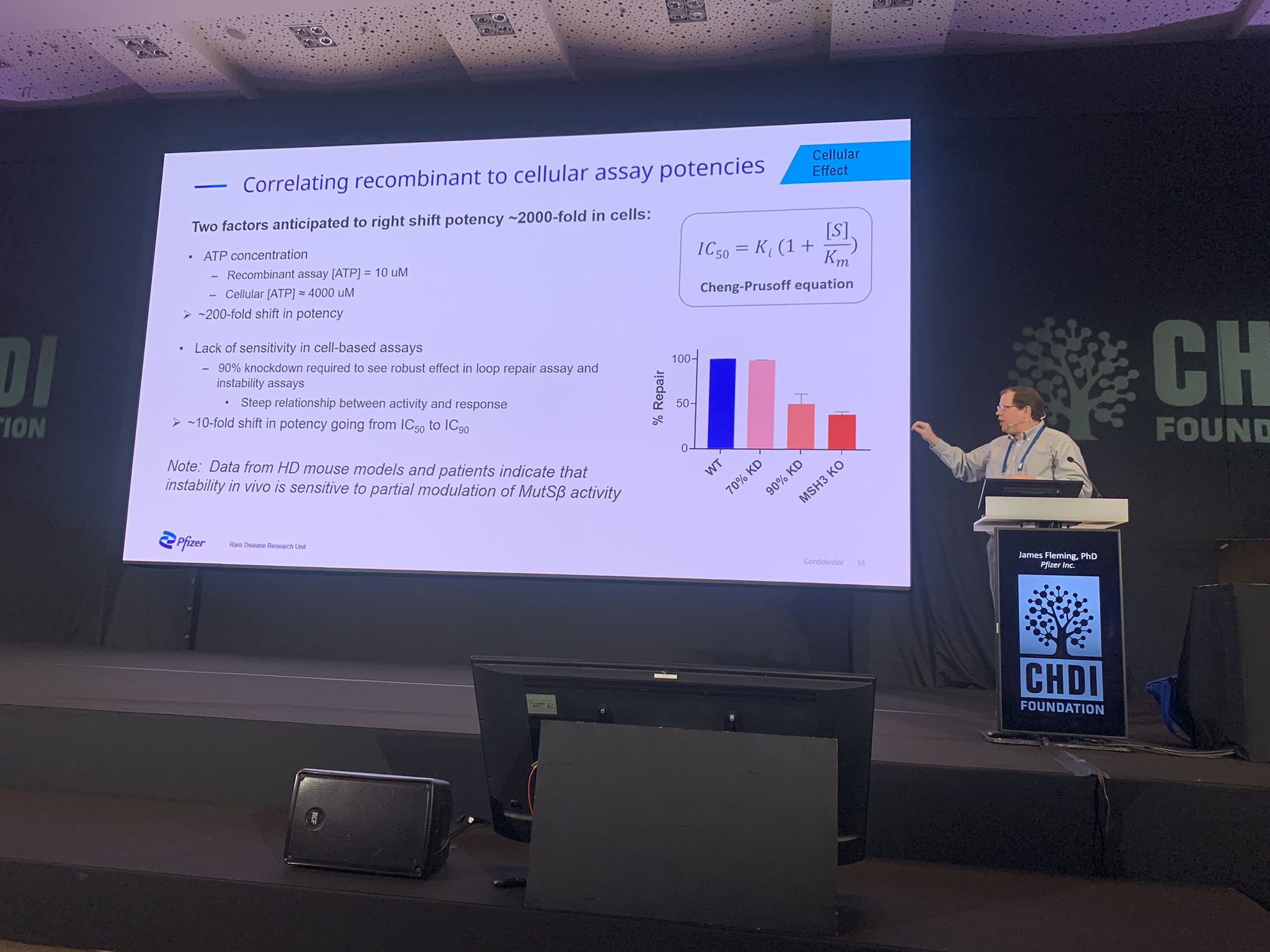
Der nächste Redner ist James Fleming von Pfizer. Auch dieses Unternehmen entwickelt Medikamente, die auf den MSH3-Stoffwechselweg abzielen. Pfizer verfolgt einen ähnlichen Ansatz wie das CHDI-Team und hat wie die anderen eine Reihe von Instrumenten und Methoden entwickelt, um ihre Moleküle auf die Fähigkeit zu testen, die Expansion von CAG-Wiederholungen zu stoppen.
Wie andere Pharmaunternehmen führt auch Pfizer eine Reihe von Schritten durch, um potenzielle Wirkstoffe zu prüfen, zu verstehen, wie sie mit den Proteinen interagieren, auf die sie abzielen, und sie dann in Zellen und an Tieren zu testen. Auch Pfizer setzt 3D-Modelle und chemische Tests ein, um zu zeigen, dass ihre Medikamente an dem Proteinkomplex haften können, zu dem MSH3 gehört, was ihnen geholfen hat, diese Moleküle mit der Zeit immer besser zu machen. Der nächste Schritt besteht darin, diese Medikamente in Zellen zu testen, die in einer Schale gezüchtet werden. Ein großer Teil dieser Arbeit konzentriert sich auf die winzigen Details der Proteinchemie, -struktur und -energetik. Es genügt zu sagen, dass Mathematik bei der Entwicklung von Medikamenten eine große Rolle spielt!
Um diese Studien an Tieren und später am Menschen durchführen zu können, wird ein Medikament mit den richtigen Eigenschaften benötigt: die Fähigkeit, auf MSH3 abzuzielen, die Fähigkeit des Körpers, es abzubauen, und die Fähigkeit des Medikaments, ins Gehirn zu gelangen. Keine leichte Aufgabe! Nachdem man die Eigenschaften eines neuen Medikaments in Zellen und Tieren besser verstanden hat, kann es dann auf seine Sicherheit bei Menschen getestet werden. Im Moment liegt dies für alle heute vorgestellten Wirkstoffe noch in weiter Ferne, aber es ist spannend zu sehen, dass die Unternehmen die Arbeit vorantreiben.
Massive Datensätze zur Identifizierung genetischer Modifikatoren
Den Auftakt der letzten Sitzung des Tages macht Jim Gusella aus Harvard, der im Namen eines großen Konsortiums von Wissenschaftlern, die sich mit der Genetik der Huntington-Krankheit befassen, über genetische Modifikatoren der Huntington-Krankheit sprechen wird.
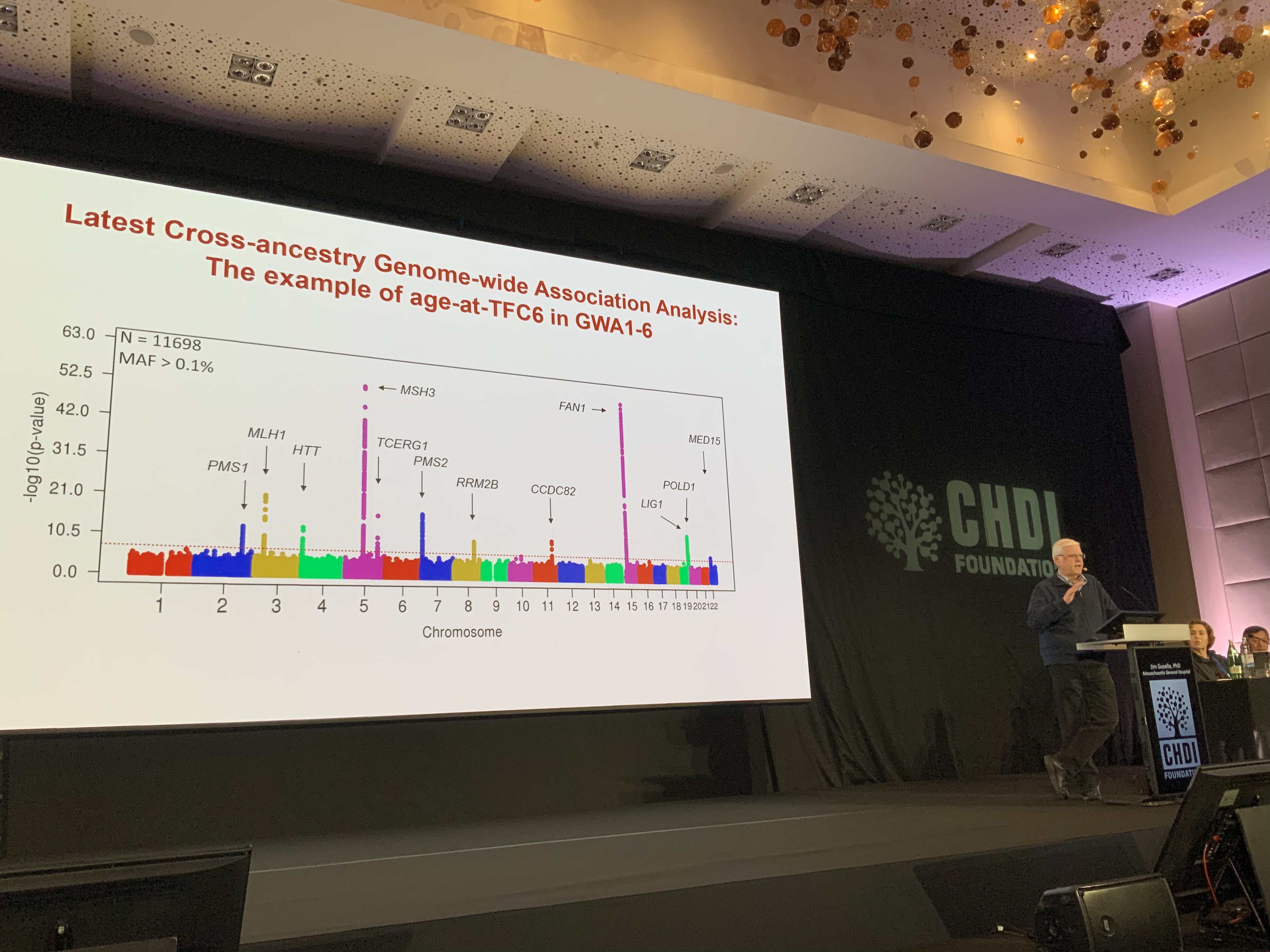
Zu Beginn seines Vortrags dankt Jim Gusella allen HD-Familien, die den HD-Forschern im Laufe der Jahre so großzügig ihre Daten und Proben zur Verfügung gestellt haben und ohne die diese groß angelegten Analysen nicht möglich wären. Eine interessante Erkenntnis, die wir schon seit einiger Zeit kennen, ist, dass bei Menschen mit der gleichen CAG-Nummer die ersten Symptome in sehr unterschiedlichem Alter auftreten können. Genetische Modifikatoren sind Marker in der DNA, die dieses frühe oder späte Auftreten der Symptome erklären können.
Immer mehr Hinweise deuten auf einen bestimmten Faktor hin, der für das Auftreten und die Geschwindigkeit, mit der sich die Huntington-Krankheit im Laufe der Zeit verschlimmert, verantwortlich ist: die Ausbreitung von CAG-Wiederholungen in einigen Zellen. Dieser Prozess, der alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. somatische Instabilität bekannt ist, scheint direkt mit genetischen Modifikatoren verbunden zu sein. Die Aussagekraft dieser Studien über Modifikatoren ergibt sich aus der Anzahl der analysierten Patientenproben – mehr Daten bedeuten eine höhere Sicherheit der Schlussfolgerungen. In der jüngsten Studie wurden die Daten von 11698 Teilnehmern analysiert, was erstaunlich ist!
Ein Problem der Huntington-Forschung und der Wissenschaft im Allgemeinen besteht darin, dass viele der analysierten Proben von Europäern oder Menschen europäischer Herkunft stammen. In diesem neuen Datensatz arbeitet das Team daran, eine vielfältigere Gruppe von Patienten in die Daten einzubeziehen. Mit einer solchen Fülle und Vielfalt an Daten ist es möglich, in großem Maßstab zu zoomen und allgemeine Vorhersagen darüber zu treffen, wie genetische Modifikatoren – winzige Veränderungen in anderen Genen – den Zeitpunkt beeinflussen, zu dem Menschen mit Huntington bestimmte Stadien der Huntington-Krankheit erreichen können.
Es ist wichtig zu betonen, dass dies ein Weg ist, um mehr Gewissheit darüber zu erlangen, welche anderen Gene bei Menschen mit Huntington die meisten Auswirkungen haben. Dies ist etwas anderes alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. eine Vorhersage des Ausbruchs oder des Krankheitsverlaufs bei einer einzelnen Person mit HK. Jims Team und das genetische Konsortium untersuchen auch, wie subtile Unterschiede und „Unterbrechungen“ in der Sequenz der CAGs im Huntingtin-Gen die DNA-Struktur und die Tendenz der Wiederholungen, instabil zu werden und länger zu werden, beeinflussen. Die gute Nachricht dieses jüngsten Datensatzes ist, dass MSH3, das Thema der vorangegangenen Sitzung, bei allen von Jim und seinen Kollegen verwendeten Analysen immer noch ein sehr bedeutender Modifikator ist. Das verleiht all diesen Ansätzen, die auf die somatische Instabilität abzielen und versuchen, die Ausdehnung der CAG-WiederholungCAG-Wiederholung Der Abschnitt der DNA am Anfang des Huntington-Gens, der die Sequenz CAG viele Male wiederholt enthält und ungewöhnlich lang ist bei den Menschen, die die Huntington-Krankheit entwickeln zu stoppen oder sie zu verkleinern, eine große Glaubwürdigkeit.
CAG-Expansionen in bestimmten Gehirnzellen
AlsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. Nächstes wird Nathaniel Heintz von der Rockefeller University über seine Arbeit zum Verständnis der Gene sprechen, die bei Huntington ein- oder ausgeschaltet werden. Das Heintz-Labor hat eine Reihe von Techniken entwickelt, die es Wissenschaftlern ermöglichen, die Kerne vieler verschiedener Zellen zu „sortieren“ und die genetischen Botschaften in vielen Zelltypen zu untersuchen. Dies ist eine wichtige Methode, um zu untersuchen, warum bestimmte Zellen bei Huntington und anderen Krankheiten besonders anfällig sind. Für diese Analysen werden postmortale Hirngewebeproben verwendet, die durch die erstaunliche Großzügigkeit der HK-Gemeinschaft ermöglicht werden.
Das Striatum, ein Bereich im Zentrum des Gehirns, ist am stärksten von HD betroffen. Heintz‘ Team ist in der Lage, verschiedene Arten von Zellen im Striatum zu untersuchen, und hat herausgefunden, dass die CAG-Repeat-Expansion am häufigsten in einer Art von Zelle, den Medium Spiny Neurons, auftritt.
Wir wissen schon seit langem, dass die mittleren Stachelneuronen (MSN) bei HD in großer Zahl verloren gehen. Es gibt verschiedene Arten von MSN, und seltsamerweise stellt sich heraus, dass sowohl die MSN, die bei der Huntington-Krankheit gefährdet sind, alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. auch diejenigen, die überleben, von der CAG-Repeat-Expansion betroffen sind. Der Grund dafür, dass diese mittelkernigen Neuronen anscheinend stärker expandieren, könnte in den höheren MSH3-Werten liegen, die in diesen Zellen zu finden sind, aber dieser Zusammenhang ist noch nicht bewiesen. Bei den mittelstacheligen Neuronen ist auch eine große Anzahl von Genen bei der Huntington-Krankheit an- oder abgeschaltet – 1.000 sind an- und 500 abgeschaltet! Viele der betroffenen Gene sind an der Reparatur von DNA-Schäden beteiligt – was wiederum die wichtige Rolle unterstreicht, die dies wahrscheinlich bei der Huntington-Krankheit spielt.
Laufende Arbeiten befassen sich mit der Frage, wann die mittleren Stachelneuronen bei Huntington betroffen sind und wie man am besten eingreift. Die Forscher untersuchen auch andere Hirnbereiche und gehen Schicht für Schicht vor, um genau zu verstehen, welche Arten von Zellen geschädigt werden oder verloren gehen.
Geschwindigkeit der CAG-Expansion
Der letzte Redner an diesem Tag ist Steve McCarroll aus Harvard. In seinem Labor wird untersucht, welche Gene auf der Ebene einzelner Zellen ein- oder ausgeschaltet werden, und nicht auf der Ebene eines großen Gemischs aus vielen verschiedenen Zelltypen – ein unglaublich detaillierter Ansatz. Er verwendet eine Fruchtanalogie, um die Leistungsfähigkeit dieser Technik zu beschreiben – man kann Zelltypen wie verschiedene Beeren vergleichen, denselben Zelltyp bei verschiedenen Menschen wie Äpfel mit Äpfeln, die Unterschiede zwischen verschiedenen Zellen desselben Typs wie bei zwei Heidelbeeren.
Anhand dieser Einzelzellanalysen können sie herausfinden, welche Zellen im Laufe der Huntington-Erkrankung verschwinden, was frühere Erkenntnisse bestätigt, wonach mittelgroße stachelige Neuronen und stachelige Projektionsneuronen die anfälligsten Arten von Nervenzellen sind. Sie können auch genau feststellen, welche Zellen CAG-Expansionen aufweisen – dies scheint darauf hinzudeuten, dass die anfälligen mittelstacheligen Neuronen am ehesten eine solche Expansion aufweisen. Die CAG-Expansion während des Lebens einer Person in diesen Zellen scheint sehr spezifisch zu sein, NUR für das Huntingtin-Gen, und nur für das HK-Gen, nicht für andere Gene, die ähnliche Arten von DNA-Code haben. Die Mehrheit dieser anfälligen Gehirnzellen weist eine moderate Expansion der CAG-Wiederholungen auf, aber eine kleine Untergruppe hat riesige Expansionen, deren Grund die Wissenschaftler noch nicht ganz herausgefunden haben.
Den Daten des McCarroll-Labors zufolge scheinen die moderaten Expansionen im Laufe der Zeit sehr langsam abzulaufen, während die übertriebenen Expansionen sehr viel schneller ablaufen. Die Schlüsselfrage lautet: Ab welchem Schwellenwert der CAG-Wiederholungszahl beschleunigt sich die Expansion und führt zur Schädigung und zum Tod dieser empfindlichen Nervenzellen? Um dies herauszufinden, kann das McCarroll-Labor einzelne Neuronen mit unterschiedlichen CAG-Zahlen vergleichen und sie in Gruppen aufteilen, um besser zu verstehen, welche Längen am problematischsten sind. Sie gruppierten die Zellen nach der CAG-Zahl und stellten merkwürdigerweise nicht allzu viele Unterschiede bei den Genen fest, die bei niedrigeren Wiederholungslängen an- und abgeschaltet werden. Die tiefgreifendsten Veränderungen treten in Zellen mit sehr, sehr langen CAG-Wiederholungen auf, mehr alsALS Eine fortschreitende Nervenkrankheit, bei der Bewegungsneuronen absterben. 180.
McCarroll schlägt eine ganz andere Sichtweise auf die Pathologie der Huntington-Krankheit und die Art und Weise vor, wie die Krankheit im Laufe der Zeit verläuft. Es gibt einige interessante Gespräche im Publikum! Aber gerade deshalb ist es so gut, dass all diese Wissenschaftler auf dieser Tagung zusammenkommen können, um all diese Ideen zu diskutieren.
Schalten Sie morgen ein!
Das war’s für heute, liebe Leute. Wir machen jetzt Schluss für heute, aber wir sind morgen früh wieder da! Vergessen Sie nicht, die Live-Updates für den Rest der Konferenz mit dem Hashtag #HDTC2023 zu verfolgen.
For more information about our disclosure policy see our FAQ…


