
Huntington's Disease Therapeutics Conference 2020 – Tag 1
HDBuzz berichtet von der jährlichen Huntington’s Disease Therapeutics Conference in Palm Springs – Tag 1


Unsere neuen Autoren Rachel Harding und Sarah Hernandez berichten von der Huntington’s Disease Therapeutics Conference – dem größen jährlichen Zusammentreffen von Huntington-Forschern.
Dienstagmorgen – Genotyp und Phenotyp
Guten Morgen aus dem sonnigen Palm Springs! Wir freuen uns hier zu sein für die 15te jährliche Konferenz. Dieses Jahr kommen zu Ed Wild und Jeff Carroll unsere neuesten Autoren Dr. Rachel Harding und Dr. Sarah Hernandez hinzu. Joel Stanton fasst ihre live Tweets zusammen zu unseren Tagesberichten.

Der erste Themenblock heißt „Genotyp und Phenotyp“ und handelt von der Übersetzung der Huntington-Mutation (Genotyp) in die Huntington-Symptome (Phenotyp). Dr. Seth Ament startet mit der Beschreibung der Arbeit seiner Gruppe, die versucht erste Veränderungen in den Gehirnen von Mäusen mit mutiertem Huntington-Gen aufzuzeichnen. Die Zellen unseres Körpers und unseres Gehirns enthalten DNA, die wiederum aus über 20.000 Gencodes aufgebaut ist. Welche Gene in einer Zelle ihre Wirkung entfalten können, bestimmt, wie diese Zelle funktioniert. Aments Labor untersucht, welche Gene sich auswirken und will verstehen, welche spezifischen Faktoren Huntington-Zellen dazu bringen, verschiedene Kombinationen von Genen wirksam werden zu lassen, in der Hoffnung, dass dies vielleicht steuerbar ist.
Zuerst beschreibt Ament seine Aufzeichnungen zu den Orten, an denen das Eiweiß Huntingtin sich an die DNA anheftet. Die offensichtlichste Weise, auf die Huntingtin bestimmen könnte, welche Gene an- oder abgeschaltet sind, ist, indem es sich direkt an die DNA hängt. Es wurde bereits herausgefunden, dass das Protein sich bei Huntington-Mäusen an andere Bereiche des Genoms anheftet als bei gewöhnlichen Mäusen. Es könnte also sein, dass ein direkter Einfluss auf die DNA genommen wird, der wichtig ist, um die Huntington-Krankheit zu verstehen.
In Huntington-Mäusen, heftet sich das mutierte Huntingtin an Stellen an die DNA an, wo viel passiert – hier werden Gene ausgelesen und verwendet. Das mutierte Huntingtin tut möglicherweise etwas bestimmtes an Stellen, wo Gene aktiv genutzt werden. Könnte das erklären, wie Zellen bei Huntington-Patienten, ihre Gene anders nutzen?
Tatsächlich befinden sich genau die Gene an den besagten Stellen, die sich in Zellen von Huntington-Mäusen verändern, es könnte also stimmen. Aments Team fand einen überraschenden Zusammenhang: sie können vorhersagen, wie aktiv oder inaktiv ein Bereich der DNA ist, je nachdem wie gut sich das Huntingtin dort anheftet.
Als nächstes stellt Ament die Erkenntnisse seiner Gruppe zu veränderten Genen im Gehirn von Huntington-Betroffenen dar. Erstaunliche neue Technologien ermöglichen es den Forschern, die Gene in einzelnen Zellen aufzuzeichnen. Aments Labor verwendet diese Methoden im Rahmen der NIH’s-BRAIN-Initiative. Sie untersuchten die Veränderungen in mehr als 13.000 individuellen Zellen aus Huntington-Maus-Gehirnen. Es gibt eine Anzahl an verschiedenen Arten von Zellen im Gehirn. Aments Ansatz ermöglicht es, die Veränderungen in jeder Art von Zelle separat aufzuzeichnen. So entsteht ein viel klareres Bild als bei einer gemischten Analyse. Die Ergebnisse ebnen den Weg zu einem viel genaueren Verständnis darüber, was genau in den Zellen passiert – was wiederum dabei behilflich sein könnte zu verstehen, wie die Zellen individuell behandelt werden könnten.
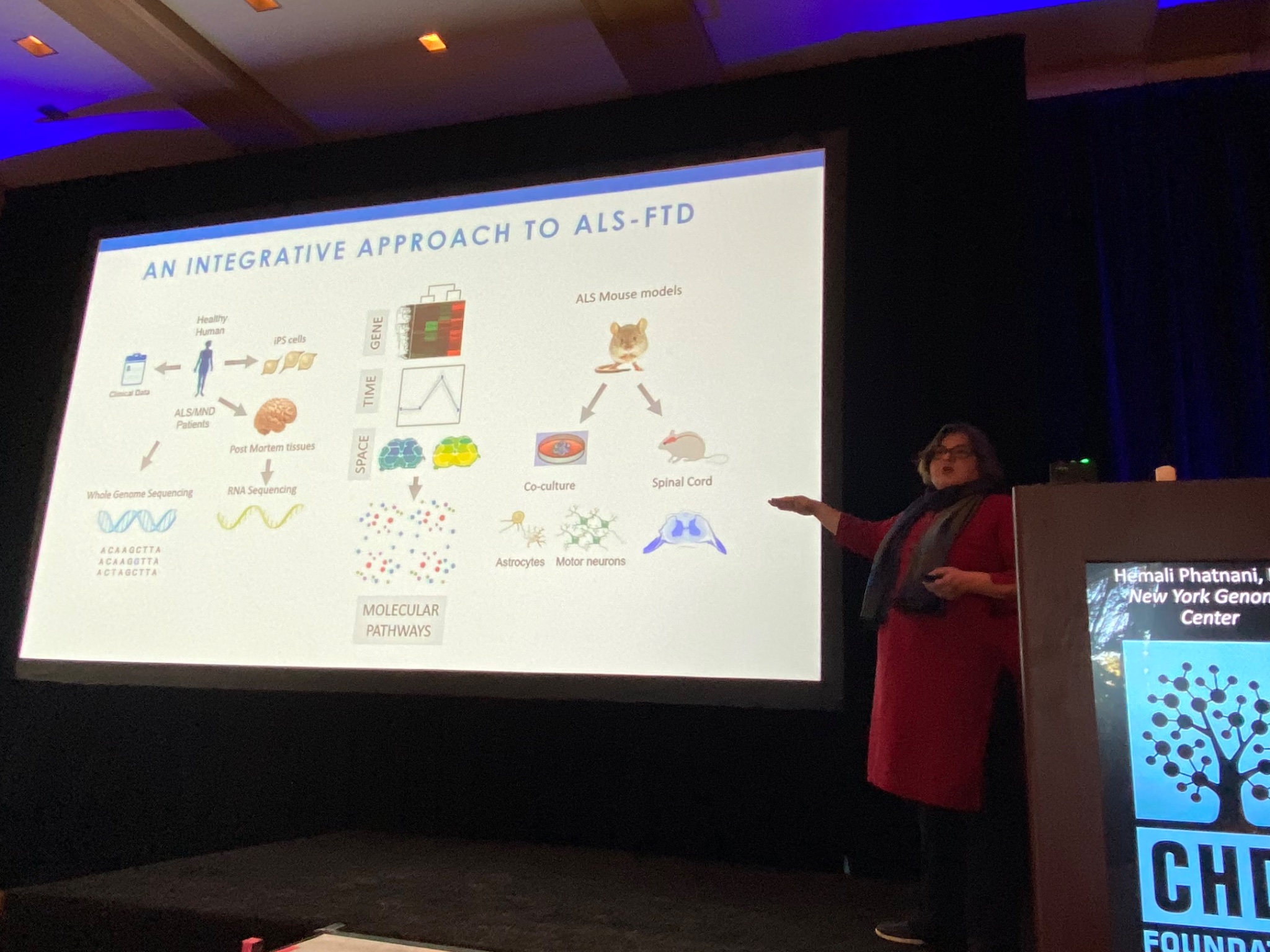
Danach ist Hemali Phatnani vom New York Genome Center an der Reihe. Sie beschreibt ihre Erforschung von Änderungen im Gehirn und im Rückenmark von Patienten mit ALS und einer weiteren verwandten Erkrankung namens FTD. Phatnani arbeitet mit einer großen Gruppe von ALS-Kliniken, um Zugang zu seltenen Proben zu bekommen, die von ALS-Patienten gespendet werden. Die Analysen dieser Proben werden als Datensätze sofort Wissenschaftlern auf der ganzen Welt zur Verfügung gestellt, ein tolles Beispiel für offene Wissenschaft. Wie die Huntington-Krankheit ist auch ALS eine komplexe Erkrankung; verschiedene Zelltypen des Gehirns erfahren unterschiedliche Schäden während ihres Verlaufs. Phatnanis Gruppe steuerte dazu bei, neue Methoden zu entwickeln, um diese Schäden aufzuzeichnen und das Zell-spezifisch. Unter folgendem Link können alle Wissenschaftler (oder neugierige Laien) auf ihre Daten zugreifen (http://als-st.nygenome.org).
Als nächstes spricht Sumanjit Jayadev vom Medical Center der University of Washington. Jayadev erforscht eine bestimmte Art von Gehirnzellen, die Mikroglia. Diese beeinflussen das Fortschreiten der Huntington-Krankheit. Das Entfernen von Mikroglia aus den Gehirnen von Huntington-Mäusen führt zur Verbesserung von Symptomen. Wissenschaftler wissen seit einiger Zeit, dass die Huntington-Krankheit eine Entzündung hervorruft und Jayadev möchte erforschen welche Zellen außer den Neuronen im Gehirn bei diesem Prozess eine Rolle spielen. Gene, die Entzündungen hervorrufen, stellen für Alzheimer-Patienten Risikofaktoren dar. Jayadev schaut sich an, in welchen Zellen des Gehirns diese Gene aktiv sind. Mithilfe ihrer Methoden kann sie Daten aus einzelnen Zellen zu diesem Zweck auswerten. Auf diese Weise wurden Unterarten von Mikroglia identifiziert, die nur bei Alzheimer-Patienten vorzufinden sind. Jayadev arbeitet mit Sage Bionetworks zusammen und stellt ihre Daten für offen für Wissenschaftler weltweit zur Verfügung. Die Daten machen es möglich den Verlauf der Alzheimer-Erkrankung gleichzeitig mit dem An- und Abschalten von Genen in individuellen Hirnzellen zu beobachten. Falls diese Technik auch für die Huntington-Krankheit angewendet werden kann, können Ärzte in der Zukunft eventuell mit der Information des Alters und der Anzahl von CAG-Wiederholungen, präzise Entscheiden, wie ein Patient am besten behandelt werden kann.
Der letzte Vortrag des Vormittags wird von William Yang gehalten. Er ist Huntington-Forscher an der University of California, Los Angeles. In seinem Labor werden große Datensätze von verschiedenen Huntington-Mausmodellen erzeugt, die Aussagen darüber treffen, wann verschiedene Gene angeschaltet werden, welche Eiweiße dann erzeugt werden und mit gesunden Mäusen verglichen werden. In den Datensätzen können Forscher nach Mustern und Zusammenhängen suchen, die eventuell anzeigen können, welche Gene bei Huntington-Mäusen miteinander wechselwirken. Diese Verhaltensmuster können mittels computergestützter Methoden aufgezeichnet werden, um zu verstehen wie bestimmte Zelltypen zu Veränderungen im Gehirn die Aktivität von Genen beeinflussen.
In seinem Vortrag konzentriert sich Yang auf die Analysen von gesunden Kontroll-Mäusen. Mit seiner Technik konnten wichtige Funktionen identifiziert werden und welche Zellen zu diesen Funktionen beitragen. Als seine Gruppe über die Auftragung der Daten von Kontroll-Mäusen, die Daten von Huntington-Mäusen legte, fanden sie heraus, dass Gene die Schlaf-Wach-Zyklen und die Reparatur von DNA regulieren sich verändert hatten. Damit bestätigen sich Ergebnisse aus anderen Forschungsarbeiten. Ähnlich könnten mit seiner Aufzeichnungstechnologie viele andere Theorien zur Huntington-Krankheit bestätigt werden – ein brauchbares Werkzeug!
Dienstagnachmittag – Somatische Instabilität
Der Nachmittagsabschnitt dreht sich um das Phänomen der somatischen Instabilität. Einfach gesagt tritt diese auf, wenn eine lange Wiederholungsfolge der DNA sich in bestimmten Zelltypen in ihrer Länge verändert.
Wir starten in den Nachmittag mit Darren Monckton von der University of Glasgow. Er interessiert sich dafür, wie somatische Instabilität die Huntington-Krankheit beschleunigt und inwiefern es eine gute Idee wäre, das Aufhalten dieser Instabilität als möglichen Behandlungsansatz zu wählen. Es ist seit einiger Zeit bekannt, dass sich die Anzahl an CAG-Wiederholungen von Gewebe zu Gewebe unterscheiden kann. Einige Zellen zeigen eine viel höhere Anzahl als andere. Das ist ähnlich wie bei anderen Krankheiten, zum Beispiel Myotone Dystrophie. Die Variationen bei der CAG-Anzahl werden stärker, wenn die Patienten älter werden, die Instabilität nimmt also zu. Weitherhin scheint bei größerer Startanzahl von CAG-Wiederholungen die Variabilität und damit die Instabilität höher zu sein. Das bedeutet nicht, dass die CAG-Anzahl bei einem einzelnen Patienten sich im Laufe des Lebens grundsätzlich erhöht, sondern nur, dass einige Zellen manchmal eine erhöhte Anzahl aufzeigen.
Die Buchstabenkombination „CAG“ steht für die Aminosäure Glutamin, daher wird die Huntington-Krankheit auch als Polyglutamin-Krankheit bezeichnet. Glutamin kann allerdings auch durch den Buchstabencode „CAA“ abgekürzt werden und obwohl die DNA dann unterschiedlich aussieht, ist das daraus resultierende Protein das gleiche, wenn ein CAG durch ein CAA ersetzt wird. Mehr Details dazu hier.
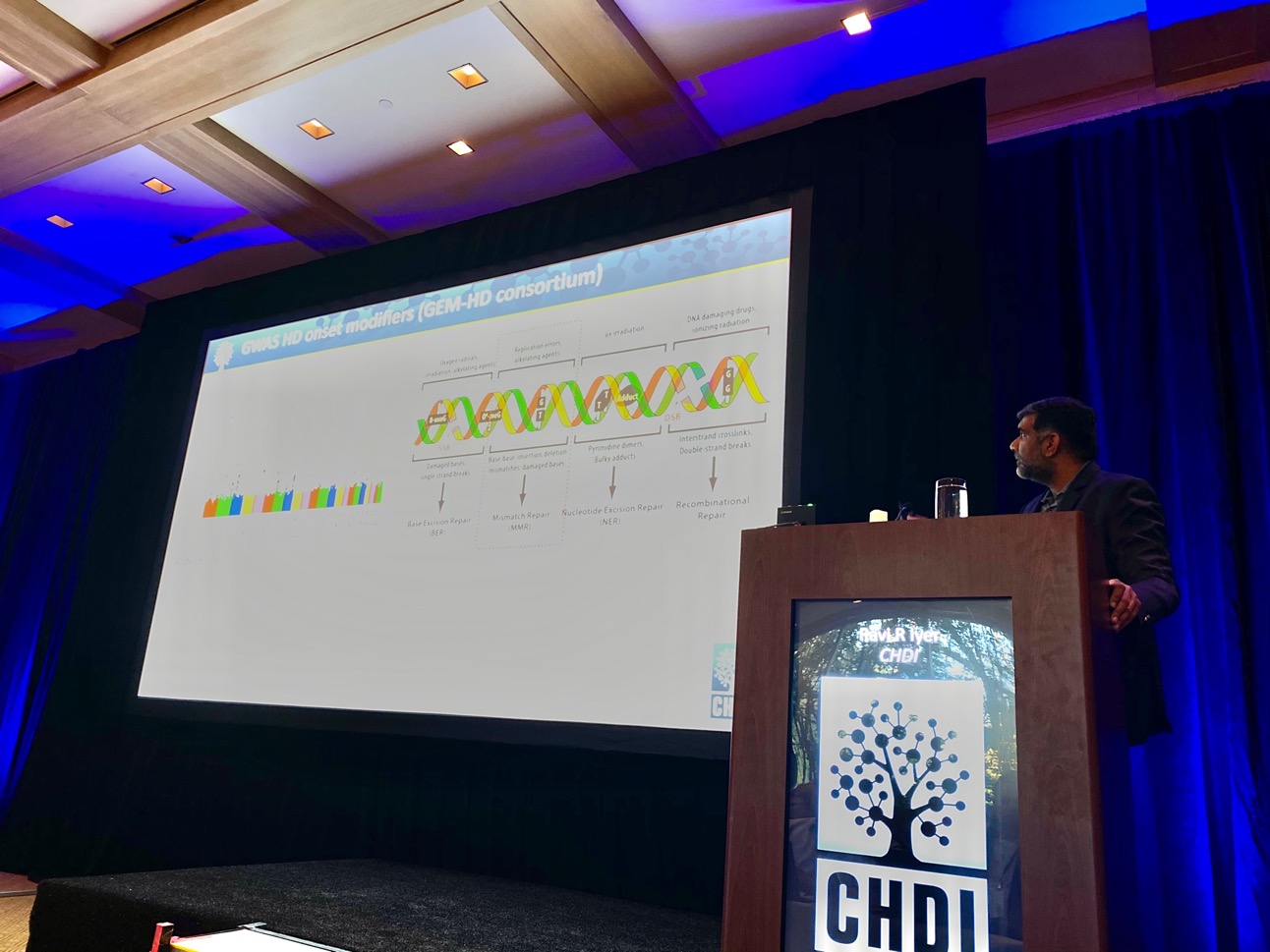
Eine kürzliche Veröffentlichung zeigt, dass die Anwesenheit einer CAA-Sequenz für eine höhere Stabilität sorgen kann. Die nächste interessante Frage ist dann, wie wird die Instabilität hervorgerufen? Forschungsergebnisse zeigen jetzt, dass die DNA-Reparatur dafür interessant sein könnte, diese wird gerade in der Huntington-Forschung heiß diskutiert.
Spezifische Treiber (Gene) der Instabilität könnten zur Behandlung der Krankheit von Interesse sein. Die Wissenschaftler arbeiten gerade daran, herauszufinden, welche dieser Treiber die wichtigsten sind und welche ein geeignetes Ziel für Behandlungsmethoden darstellen können. Sie untersuchen zunächst, wie sich die Instabilität der CAG-Anzahl über die Lebensdauer eines Patienten hinweg entwickelt. Hierbei greifen sie aus Daten der Studie EnrollHD zurück.
Wenn man das Fortschreiten der Huntington-Krankheit beobachten und nachverfolgen könnte, indem man die Instabilität mit einem einfachen Bluttest erfasst, könnten Ärzte auf wenig invasive Weise den Zustand ihrer Patienten erkennen und die bestmögliche Behandlung ermitteln.
Als nächste trägt Karen Usdin von National Institutes of Health (NIH) vor. Sie hat sich auch mit somatischer Instabilität beschäftigt und zwar an Hand von Mäusen mit einer anderen Nervenkrankheit namens Fragiles-X-Syndrom. Wie bei der Huntington-Krankheit, liegt auch beim Fragilen-X-Syndrom eine sich ungewöhnlich häufig wiederholende Sequenz in einem Gen vor. Es ist in diesem Fall aber nicht die Abfolge CAG, sondern die Abfolge CGG. Die Wissenschaft geht davon aus, dass bei solch ähnlichen Ursachen auch ähnliche Krankheitsmechanismen vorliegen. Wie bei der Huntington-Krankheit gibt es auch beim Fragilen-X-Syndrom somatische Instabilität und Gene, die bei der DNA-Reparatur mitwirken, beeinflussen diese Instabilität. Usdin stellt bei den von ihr untersuchten Mäusen fest, dass das Verändern der Anzahl von DNA-Reparatur-Genen die weitere Verlängerung des CGG-Strangs verhindert und sogar einige CGG-Wiederholungen wegnimmt. Für uns ist es furchtbar interessant von solchen Erforschungen außerhalb der Huntington-Welt zu hören, die neue Ideen und Wissen einbringen und damit vielleicht die Forschung an der Huntington-Krankheit beschleunigen können!
Den folgenden Vortrag hält Ravi Iyer von CHDI. Er handelt von einem Medikamentenprogramm. Ziel des Programms ist es, kleine Moleküle zu identifizieren, die die Länge des CAG-Stranges stabilisieren. Viele Unternehmen kooperieren hier mit CHDI. Eine ihrer Methoden ist es, sich die Strukturen der Zielmoleküle genau anzusehen, beispielsweise Proteine, die bei der DNA-Reparatur mitwirken. Der größte Vorteil von kleinen Molekülen ist, dass sie in Form einer Tablette eingenommen werden könnten. Daher handelt es sich um einen spannenden Ansatz. Gleichzeitig müssen die Forscher hier aber besonders vorsichtig sein, damit keine unerwünschten Nebenwirkungen hervorgerufen werden. Man muss noch einen weiten Weg zurücklegen, bevor wir sagen können, ob kleine Moleküle einen Erfolg haben werden. Es ist eine große Gruppe von Wissenschaftlern, die sich angeführt von CHDI aktuell damit beschäftigen und wir freuen uns auf Neuigkeiten bei künftigen Konferenzen.
Anschließend berichtet Brian Battencourt für Triplet Therapeutics, einer Firma, die im oben beschriebenen Projekt arbeitet. Er soll mehr über die Therapiemöglichkeiten von somatischer Instabilität erklären. Eines der Ziele ist, die zusätzliche Verlängerung der CAG-Wiederholungssequenz aufzuhalten und den Ausbruch der Huntington-Krankheit herauszuzögern, am besten bis zu einem Alter, das realistischerweise gar nicht erreicht wird. Die Wissenschaftler legen Prioritäten fest, welche Moleküle sie als erstes untersuchen. So können sie effizient an ihrem Ziel arbeiten, potentielle Medikamente so schnell wie möglich zu entdecken. Nachdem sie zunächst auf die Sicherheit und niedriges Gefahrenpotential geschaut hatten, entwickelte Bettencourts Gruppe Moleküle, die 8 verschiedene Gene ansteuern. Das ist zwar eine große Anzahl von Anknüpfungspunkten, sie ist aber noch klein genug um relativ schnell abgearbeitet zu werden. Wie beschrieben ist diese Forschung Teil eines größeren Konsortiums und Bettencourt arbeitet z. B. mit HDBuzz-Autor Jeff Carroll zusammen. Wir gehen davon aus, dass sie in der Lage sein werden, im nächsten Jahr näheres zu berichten.
Den letzten Vortrag des Tages hält Jeff Carroll, der die zweite Tageshälfte zum Thema somatische Instabilität zusammenfasst. Eines der Themen, für die er sich interessiert, ist die Auswirkung von Huntingtin-Verminderung außerhalb des Gehirns, beispielsweise in der Leber. Interessanterweise führt die Verminderung von Huntingtin zu einem Absenken in spezifischen Gewebearten, in anderen dagegen nicht. Jeff Carrolls Arbeitsgruppe wollte das genauer verstehen, daher arbeiten sie zusammen mit Prof. Sarah Tabrizi an der Forschung an menschlichen Neuronen.
In einem Ataxie-Mausmodell (Ataxie ist eine andere Erkrankung, die auf zu viele CAG-Wiederholungen zurückzuführen ist) wurde bei Huntingtin-Verminderung festgestellt, dass sich auch die somatische Instabilität reduzierte. Dies könnte bedeuten, dass das mutierte Huntingtin selbst ein Eiweiß ist, dass bei zusätzlichen Verlängerung eine Rolle spielt. Falls das so ist und falls Huntingtin beeinflusst, wie gut unsere DNA erhalten bleibt, möchte die Gruppe von Carroll herausfinden, wie genau das vor sich geht und was die Auswirkungen für Huntington-Patienten sind. Es handelt sich hierbei noch um eine sehr neue Beobachtung.
Das wär’s vom ersten Tag der Konferenz, wir berichten bald auch von Tag 2 und 3!
Weitere Informationen zu unseren Offenlegungsrichtlinien finden Sie in unseren FAQ…


