
Huntington's Disease Therapeutics Conference 2019 – Tag 3
Neue Details zum Huntingtin-Eiweiß und weitere Fortschritte bei der Entwicklung von Behandlungsmethoden mithilfe genetischer Besonderheiten: Huntington's Disease Therapeutics Conference 2019 – Tag 3

Dr. Jeff Carroll und Dr. Ed Wild berichten von der “Huntington’s Disease Therapeutics Conference” – der größten jährlichen Zusammenkunft von Huntington-Forschern. Die Konferenz in diesem Jahr ist noch größer und aufregender als zuvor.
Lesen Sie über Tag 1 hier.
Lesen Sie über Tag 2 hier.

Donnerstagmorgen – Huntingtin-Struktur und -Funktion
Der letzte Tag der Konferenz beginnt mit einem Abschnitt zur Biologie des Huntington-Gens selbst. Es ist wichtig, zu verstehen, was dieses Gen normalerweise tut und wie seine Funktionen durch die Mutation, die die Huntington-Krankheit auslösen, verändert werden. Jeder Mensch hat das Huntington-Gen. Wozu ist es also da und was tut es, um unseren Zellen beim Überleben zu helfen?
Zuerst spricht Rick Myers von der Firma Hudson Alpha, der beschreibt, dass in seinem Labor daran gearbeitet wird, zu verstehen, wodurch das Huntington-Gen angeschaltet wird. Unsere Gene befinden sich in unserer DNA und bleiben inaktiv, bis sie von bestimmtne Zellprozessen aktiviert werden. Myers Arbeit könnte dabei helfen, die grundlegende Biologie des Huntington-Gens zu verstehen, könnte dann aber auch dazu beitragen, neue Werkzeuge für die Unterdrückung seiner Funktion zu entwickeln. Das Huntington-Gen befindet sich nicht nur in Gehirnzellen sondern in vielen anderen – vielleicht in allen – Zellen des Körpers.
Myers ist Teil eines großen Forscherkonsortiums, dem sogenannten ENCODE-Project, das sich damit beschäftigt, wie bestimmte Veränderungen in der Struktur der DNA, bestimmte Gene befähigen, an- und ausgeschaltet zu werden. Das ENCODE-Projekt hat eine eigene Wikipedia-Seite.
In den Zellen wird die DNA chemisch verändert, um sie fester und dichter oder lockerer und offener zu machen. Sie bildet auch Schlaufen – Myers beschreibt die Wechselwirkung von DNA-Schlaufen über lange Distanzen. Myers Team hat DNA-Abschnitte identifiziert, die über 100.000 DNA-Buchstaben entfernt vom Huntington-Gen selbst, dessen Wirkung bestimmen. Die Forscher erstellen sozusagen eine Landkarte der DNA-Nachbarschaft um das Huntington-Gen – wie wird es angeschaltet, wie wird es abgeschaltet? Gibt es einen Zusammenhang mit den großen DNA-Schlaufen in der Umgebung?
Als nächste am Rednerpult ist Professor Gill Bates vom University College London. Sie beschreibt die Arbeit ihres Labors zum Verständnis des „Spleißens“ des Huntington-Gens. Die meisten Gene – insbesondere die größeren wie das Huntington-Gen – werden aus Stücken von DNA geschrieben, die von langen Passagen von DNA durchsetzt sind, die allerdings nicht zum Aufbau der Proteine beitragen. Das heißt die meiste DNA am Ort des Huntington-Gens ist eigentlich gar nicht Bestandteil des Gens. Seltsam diese Biologie! Wenn Zellen die Information des Gens auslesen sollen, muss jedes relevante Stück gelesen werden, wie die Kapitel eines Buches. Alle unwichtigen Passagen müssen ausgelassen werden. Das Huntington-Gen besteht aus 67 relevanten Kapiteln. Diese Kapitel zusammenzufügen bezeichnet man als „spleißen“.
In Bates Labor wurde etwas Kurioses entdeckt: bei mutierten Huntington-Genen bleibt beim Spleißen zwischen Kapitel 1 und Kapitel 2 manchmal ein kleines Stück überflüssige DNA erhalten. Das sollte nicht passieren – im Normalfall wird jedes Kapitel exakt erfasst und an seine Nachbarn angehängt. Die Folge der zusätzlichen DNA ist die Herstellung eines winzigen Proteins, das etwa 3 % des Huntingtin-Proteins ausmacht. Was dieses Protein in den Gehirnen von Menschen mit mutiertem Huntington-Gen auslöst, will Bates Labor untersuchen.
Momentan betrachten sie gespendete Hirnproben von Betroffenen. Hier finden sie Nachweise für die Abweichungen beim Spleißen, die sie vorher bereits bei Mäusen beobachtet hatten. Spielen diese Abweichungen eine Rolle? Bates Labor arbeitet mit zwei Stämmen von Huntington-Mäusen, die unterschiedliche Schweregrade der Krankheit in sich tragen, und versucht so, die Auswirkungen zu verstehen. Es zeigt sich, dass Mäuse, bei denen die Veränderung beim Spleißen auftritt, stärker erkranken, was sich mit der Hypothese der Gruppe deckt. Ganz neu arbeitet Bates Labor an Mäusen, bei denen das Extra-Stück DNA, das versehentlich beim Spleißen erhalten bleibt, von Anfang an nicht existiert. Bei diesen Mäusen wird das winzige Protein nicht hergestellt. Sie sollten es also ermöglichen, Rückschlüsse auf den Einfluss des Spleißfehlers auf den Verlauf der Huntington-Krankheit bei Mäusen zu ziehen. Was das Ganze dann für menschliche Betroffene bedeutet, muss noch erforscht werden.
Als Nächster spricht Chris Ross von der Johns Hopkins Universität über „post-translatorische Modifikation“ – dabei geht es um kleine chemische Veränderungen, die das Verhalten von Proteinen beeinflussen. So in etwa, wie eine ärodynamische Form ein Auto schneller machen kann. Das Eiweiß Huntingtin kann auf viele Arten modifiziert werden, so viel ist bekannt. Man weiß auch, wo sich diese Modifikationen entlang der Molekülketten befinden, allerdings nicht, was durch sie ausgelöst wird und warum sie dort sind. Daher kann man auch nicht sagen, ob sie das Protein harmloser oder schädlicher machen.
Ross’s Team schaute sich große Zahlen individueller Modifikationen an und testete sie in Zellkulturen. Sie untersuchten, ob das Protein die Zellen schneller oder weniger schnell zerstört und fanden heraus, dass manche Veränderungen die Schäden durch das Protein eindämmten. Einige dieser Modifikationen und die Enzyme, die sie vornehmen, könnten also gute Ansätze für die Entwicklung neuer Medikamente darstellen.

Ein großer Durchbruch gelang im letzten Jahr mit der Entschlüsselung der Struktur des Huntingtins als dreidimensionales Gebilde – dazu können Sie hier mehr lesen. Dadurch können Forscher wie Ross nun zum ersten Mal verstehen, wo genau sich die Modifikationen auch in räumlicher Hinsicht befinden. Aber wie wird aus diesen Modifikationen nun ein Medikament? Als erstes ist herauszufinden, welche Modifikation wünschenswert ist (das Huntingtin also weniger schädlich macht). Danach muss erfasst werden, welches Enzym diese Modifikation hervorruft.
An dieser Stelle lief ein beeindrucktes Raunen durch die Zuhörerschaft, da die vorgetragene Arbeit zwei wissenschaftliche Bereiche – Struktur des Eiweißes und translatorische Modifikation – so gewinnbringend verknüpft.
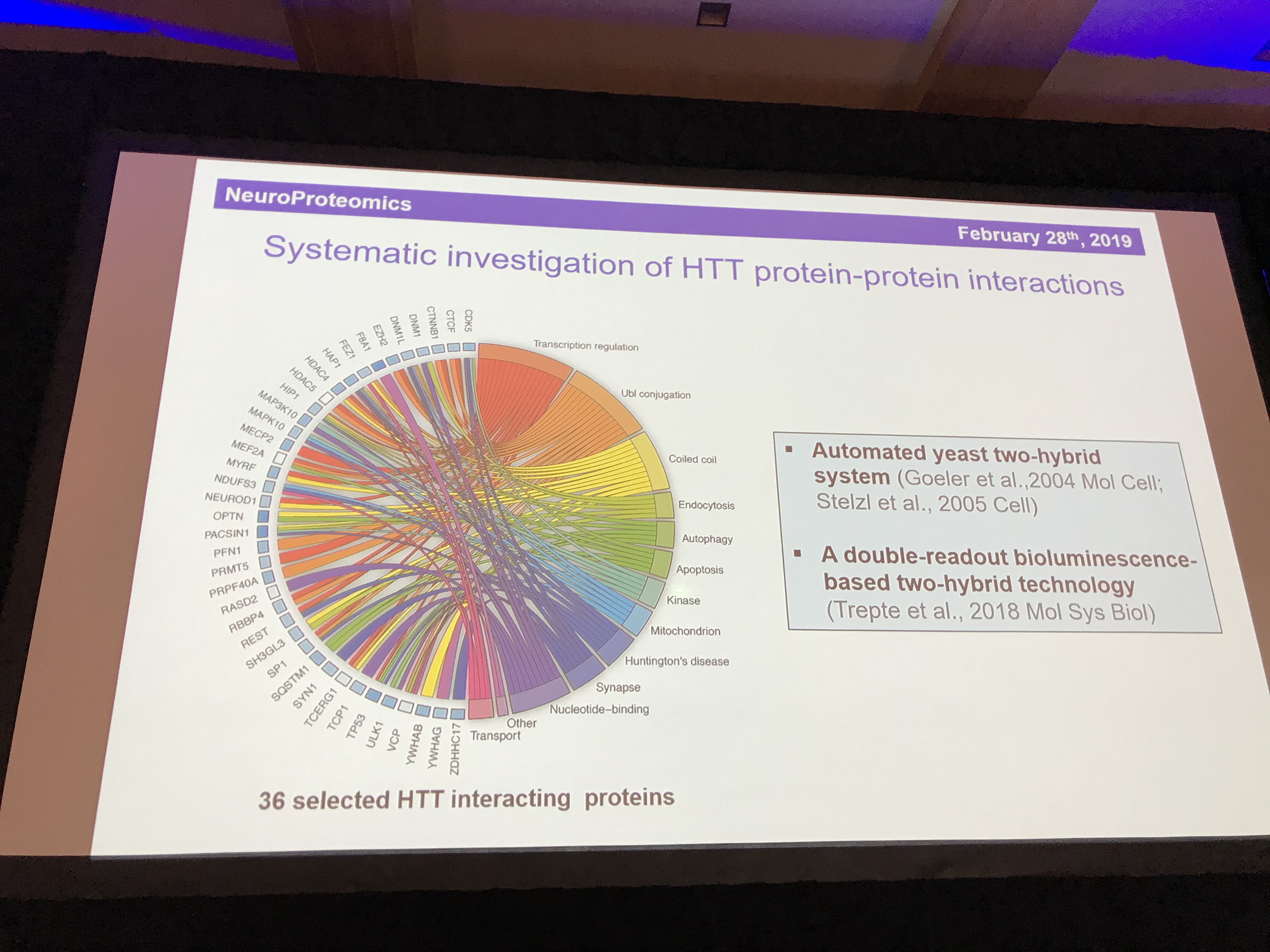
Der letzte Sprecher dieses Abschnitts ist der österreichische Forscher Erich Wanker. Er ist ein Experte in Sachen Verhalten des Huntingtins. Im Labor verwendet er Fruchtfliegen, um zu studieren, wie das mutierte Huntingtin das Absterben von Zellen verursacht. Auch seine Arbeit wurde durch die genaue Erfassung der Struktur des Proteins einen entscheidenden Schritt vorangebracht. Weiterhin ist eine der wichtigsten Informationen über ein Eiweiß, mit welchen anderen Eiweißen es sich umgibt. Zwischen benachbarten Eiweißen gibt es Wechselwirkungen, sie werden auch „Interaktionspartner“ genannt.
Aus 413 möglichen Interaktionspartnern konnte Wanker 36 herausfiltern, die interessant sein könnten. Gerade ist der dabei die in der Literatur beschriebenen Interaktionen in seinem Labor nachzuprüfen und dabei herauszufinden, wie stark das Huntingtin interagiert. Dazu forscht er an lebenden Zellkulturen. Sein Testsystem erfasst die Stärke und Nähe der Interaktionen, um herauszufinden, welche Partner für das Huntingtin am wichtigsten sind. Diese deuten auf Funktionen des Huntingtins im Bereich der Verwertung, des Transports und der Verpackung von Signalmolekülen in kleine Bläschen names Vesikel hin. Die Wirkungen der Interaktionen zu studieren, kann also dabei helfen, die Rolle von Huntingtin sowohl in seiner wilden als auch in seiner mutierten Form zu verstehen.
Donnerstagnachmittag – DNA-Reparatur und -Handling
Zum Abschluss der Konferenz gibt es noch einen aufregenden Abschnitt zur Einflussnahme anderer Gene – außerhalb des Huntington-Gens – die dennoch für die Huntington-Krankheit eine wichtige Rolle spielen. Es wurden bereits Variationen von diesen Genen entdeckt, die den Ausbruch der Krankheit früher oder später im Leben stattfinden lassen können. Auf HDBuzz finden Sie näheres dazu hier.
Als erste spricht Marcy MacDonald von Massachusetts General, eine Schlüsselfigur in jener Gruppe, die 1993 das Huntington-Gen identifizierte. Seit damals hat sie immer weiter am Verständnis der Genetik hinter der Huntington-Krankheit gearbeitet.
MacDonald stellt die Arbeit des so genannten „GeM-HD“-Konsortiums vor. Das Konsortium beschäftigt sich mit den eigangs beschriebenen Genen und deren Variationen. Sie sagt: „Die Natur hat bereits eine erfolgreiche Studie zur Huntington-Krankheit durchgeführt“, und meint damit, dass der Einfluss diverser Gene und ihrer Variationen an realen Betroffenen beobachtet werden kann. Die Daten aus beobachtenden wissenschaftlichen Studien zeigen bereits, dass eine positive Einflussnahme möglich sei, man brauche sich das also nicht mehr zu fragen. Man müsse nur herausarbeiten, wie diese genetischen Effekte künstlich reproduziert werden können, sodass man eine Therapie daraus machen kann.
Die Studien zu genetischen Einflussnehmern beginnen also bei der Sammlung von Daten vieler Huntington-Patienten, bei denen das Alter, in dem die Krankheit ausgebrochen ist, bekannt ist. Auch wenn dieses Alter stark mit der Anzahl der CAG-Wiederholungen im mutierten Gen zusammenhängt, gibt es trotzdem einen großen Streubereich, sodass es auch andere Einflüsse geben muss. Mithilfe von speziellen Mikrochips können die Forscher riesige Mengen an DNA-Buchstaben auslesen – etwa 11 Million. So kann eine detaillierte Karte des Genoms eines jeden Teilnehmers erstellt werden.
Nachdem das bei 4.000 Patienten vorgenommen wurde, identifizierte das GeM-HD-Konsortium vier verschieden Regionen des Genoms, die bei Menschen mit frühem oder spätem Ausbruch gefunden wurden. Tatsächlich konnten sie Variationen feststellen, die den Ausbruch entweder beschleunigten oder verzögerten. Betroffene, die solche Variationen haben, könnten also eventuell eine genaure Aussage über den Ausbruchszeitpunkt der Krankheit erhalten. Weiterhin lernen wir daraus viel über die Krankheit selbst. Interessanterweise sind einige der identifizierten Gene nämlich wichtig für einen bestimmten Prozess: die Reparatur von DNA. Das legt also nahe, dass eine gestörte DNA-Reparatur den Verlauf der Krankheit negativ beeinflusst.
MacDonald stellt die Ergebnisse ihrer aktuellen Studie mit mehr als 9.000 Teilnehmern vor. Hierbei zeigen sich einige weitere Gene, die den Ausbruchszeitpunkt der Huntington-Krankheit bestimmen, darunter sind noch einige DNA-Reparatur-Gene. Interessanterweise zeigten sich auch seltene Variationen im Huntington-Gen selbst als Modifikatoren hinsichtlich des Ausbruchs. Die Hintergründe sind etwas komplexer, wir werden daher in Kürze einen weiteren HDBuzz-Artikel dazu veröffentlichen.
MacDonald’s Team entwickelt auch mathematische Techniken, die das Fortschreiten der Huntington-Krankheit besser verständlich machen sollen. Sie wissen, dass das komplizierter ist als die bloße Diagnose. Insbesondere haben sie versucht, abzuschätzen, wie beeinträchtigt die Alltagsfähigkeiten von betroffenen Personen im Verlauf der Krankheit sind, anstelle von der Angabe eines einzigen fixen Zeitpunktes der Diagnose oder des Ausbruchs der Krankheit. Mithilfe der neuen mathematischen Modelle werden die vorangeganenen Studien zu genetischen Modifikatoren nun erneut analysiert, um zu sehen, welche Gene für welche Teilaspekte der Krankheit von Bedeutung sind.
Als nächster kommt Peter Holmans von der Cardiff University. Auch er ist ein Mitglied des GeM-HD-Konsortiums und arbeitet gerade an dem Verständnis des Einflusses genetischer Veränderungen auf die Huntington-Krankheit. Auf der Konferenz spricht er über eine Art genetischen Punktwert den „polygenetischen Risikowert“. Das klingt kompliziert, aber das Konzept ist recht einfach. Es handelt sich um eine Summe kleiner genetischer Risiken, die einen Gesamtrisikowert ergeben. Anders als die Huntington-Krankheit werden viele Krankheiten nicht durch eine einzige genetische Mutation hervorgerufen, sondern entstehen aus vielen kleinen „genetischen Risiken“. Jetzt, da genetische Variationen erfasst und computergestützt verarbeitet werden können, kann ein polygenetischer Risikowert relativ einfach berechnet werden.
Holmans konzentriert sich auf die psychologischen Symptome der Huntington-Krankheit – also Aggressionen, Depressionen und Verhaltensstörungen. Seine Gruppe hat einen interessanten Ansatz, um nach genetischen Variationen zu suchen, die diese Symptome unabhängig vom allgemeinen Fortschreiten der Huntington-Krankheit beeinflussen. Polygenetische Risikowerte für psychologische Symptome wie schwere Depressionen halfen Holmans dabei festzustellen, dass Huntington-Patienten mit bestimmten genetischen Variationen eine höhere Wahrscheinlichkeit haben, solche psychologischen Symptome zu entwickeln. Im weiteren Verlauf der Forschung mag es möglich sein, dass Huntington-Familien in der Lage sind eine Vorstellung von den Symptomen zu entwickeln, für die ihr Risiko besonders hoch ist. Da viele dieser Symptome – beispielsweise Depressionen – behandelbar sind, handelt es sich hierbei um eine nützliche Information für diese Familien.
Als nächste spricht Hilary Wilkinson von der CHDI Foundation. Sie beschreibt die Anstrengungen ihrer Stiftung zu verstehen, welche Rolle DNA-Reparaturgene bei der Huntington-Krankheit spielen. Neben dem Huntington-Gen selbst, sind diese Gene aktuell die bedeutendsten möglichen Einflussnehmer auf die Huntington-Krankheit. Das rührt daher, dass Studien zu den genetischen Modifikatoren bei Huntington-Patienten nahelegen, dass winzige Veränderungen in den Abfolgen dieser Gene den Ausbruch der Huntington-Krankheit maßgeblich beeinflussen können. Der ganze Sinn hinter der Kartierung vollständiger Genome besteht darin, herauszufinden, welche Gene als Ziele für chemische Veränderungen geeignet scheinen, um wünschenswerte Geneffekte zu replizieren.
CHDI unterstützt eine Menge an Anstrengungen, um genau zu verstehen, wie DNA-Reparaturgene die Huntington-Krankheit beeinflussen und ob Medikamente entwickelt werden können, um positive Einflüsse bei Huntington-Patienten nachzubauen. Mithilfe von Maus-Modellen sind Huntington-Forscher in der Lage, präzise Genbearbeitung vorzunehmen, um die DNA-Reparatur auszuschalten und so nachzuvollziehen, wie diese das Huntington-Gen und die Huntington-Symptome beeinflusst. Wilkinson beschreibt ein Phänomen namens „somatische Instabilität“, das zu einer Verlängerung der CAG-Wiederholung im Huntington-Gen führt. Dieses tritt in einigen, aber nicht in allen, Körperzellen auf. Es tritt auch in den Gehirnzellen auf, die verursacht durch die Huntington-Krankheit, absterben. Die meisten Gene, die in diesen Genom-weiten Untersuchungen entdeckt worden, spielen bei der somatischen Instabilität eine Rolle. Daher gibt es in der Huntington-Forschung gerade ein großes Interesse an diesem Prozess.
Wilkinson schloss ihren Vortrag ab, indem sie breit angelegte Experimente durch CHDI und von akademischen Partnern beschrieb. Ziel der Experimente ist die Suche nach einem Medikament, dass den Prozess der somatischen Instabilität positiv beeinflussen kann.
Das wäre alles von der diesjähringen Huntington’s Disease Therapeutics Conference! Wie bereits erwähnt, können Sie die Tage 1 und Tag 2 hier nachlesen oder uns auf Twitter folgen.
Weitere Informationen zu unseren Offenlegungsrichtlinien finden Sie in unseren FAQ…


