Neue Studie beleuchtet regionale Degeneration bei HD
Warum sterben bestimmte Gehirnzellen bei HD ab? Neue Forschungsergebnisse deuten darauf hin, dass dies an der Effizienz des zellulären Recyclings liegen könnte
Bestimmte Hirnregionen degenerieren bei der Huntington-Krankheit schneller als andere. Steven Finkbeiners Team kommt zu dem Schluss, dass diese Diskrepanz auf unterschiedlichen Fähigkeiten der Gehirnzellen in diesen Regionen beruht, mutiertes HD-Protein schnell zu identifizieren und zu entsorgen. Insbesondere Neuronen aus anfälligen Hirnregionen sind am langsamsten darin, das Protein zu beseitigen.
Wissen, wann man sie faltet
Proteine sind große biologische Moleküle, die verschiedene essentielle Aufgaben für die Zelle erfüllen. Sobald ein Protein durch Aneinanderreihen von Aminosäuren in einer bestimmten Reihenfolge hergestellt wurde, faltet es sich wie eine Brezel zu einer einzigartigen dreidimensionalen Form. Nur wenn das Protein korrekt gefaltet ist, kann es seine normale Aufgabe erfüllen.
Leider werden einige Proteine auf dem Weg verändert, sodass sie sich nicht mehr korrekt falten. Bei HD bewirkt eine genetische Mutation, dass eine der Aminosäuren in der Kette des Huntingtin-Proteins (Htt) immer wieder wiederholt wird, wie eine springende Schallplatte (oder ein Stottern, falls Schallplatten vor deiner Zeit waren).
Diese Expansion führt dazu, dass sich Htt falsch faltet, für die Zelle toxisch wird und sich zu großen Klumpen zusammenballt, die Wissenschaftler als Aggregate bezeichnen. Ein passendes Bild wären Haarbälle, die einen Abfluss verstopfen: lose, einzelne Haare sind in Ordnung, aber die großen Klumpen verstopfen ihn.
Bei Patienten mit HD scheint jede Körperzelle mutiertes HD-Protein zu enthalten, aber es sind die Gehirnzellen, die während der Krankheit bevorzugt absterben. Deshalb ist HD eine „neurodegenerative“ Erkrankung. Tatsächlich sterben nicht nur Gehirnzellen ab, sondern innerhalb des Gehirns sterben bestimmte Neuronpopulationen am frühesten und scheinen daher am anfälligsten zu sein.
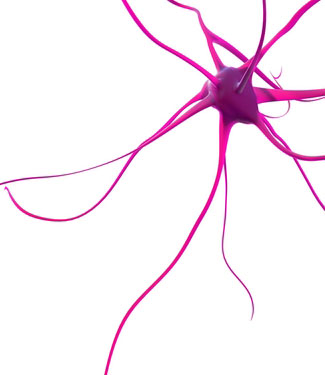
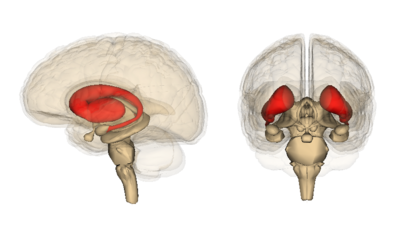
Insbesondere die Hirnregion, die den frühesten und schwersten Zelltod aufweist, wird Striatum genannt. Das Striatum liegt tief im Zentrum des Gehirns. Seine Aufgabe ist es, willkürliche Bewegungen, Gedanken und soziale Interaktionen zu koordinieren und sicherzustellen, dass die Dinge nicht außer Kontrolle geraten.
Alles, vom Beenden eines Gesprächs bis zum Stoppen einer Bewegung, wird vom Striatum gesteuert. Was extrem wichtig, aber derzeit noch nicht verstanden wird, ist, warum dieser Bereich bei HD so anfällig für Degeneration ist, während andere Regionen mit mutiertem Htt viel länger zurechtzukommen scheinen, bevor sie absterben.
„Neuronen, die mit zusätzlichem Nrf2 überladen waren, beseitigten mutiertes Htt schneller als normal und starben seltener ab. Dies deutet darauf hin, dass die Ankurbelung des zelleigenen Recyclingwegs ein potenzielles therapeutisches Ziel bei HD sein könnte.“
Papiere, Müll und mutiertes Huntingtin entsorgen
Zwei mögliche Erklärungen für diese Unterschiede wurden von einem Forschungsteam unter der Leitung von Dr. Steven Finkbeiner an der University of California in San Francisco untersucht. Eine Möglichkeit ist, dass es eine Variabilität zwischen den Gehirnzelltypen in der Geschwindigkeit geben könnte, mit der mutiertes Htt toxische Strukturen bildet. Im Grunde: Wie schnell werden Haare zu Haarbällen, für jeden Zelltyp?
Eine weitere mögliche Erklärung für den selektiven Zellverlust ist, dass verschiedene Hirnregionen mutiertes Htt unterschiedlich beseitigen und entsorgen könnten. Sind manche Zellen einfach nur Putzteufel?
Die Autoren entwickelten eine Technik, mit der sie diese Faltungs- und Beseitigungseigenschaften in einzelnen lebenden kultivierten Gehirnzellen messen konnten. Sie füllten die Zellen mit einem leuchtenden Protein namens „Dendra2“. Dieses Protein ist clever, weil es zunächst grün leuchtet (wie ein Leuchtstab-Spielzeug für Kinder), aber zu einem rotfarbenen Licht wechselt, wenn es mit einer bestimmten Lichtfarbe beleuchtet wird.
Dieses Werkzeug ermöglicht es dir also, Neuronen voller grünem Dendra2 zu züchten und sie dann mit einem Licht zu bestrahlen, das das Dendra2 rot färbt. Wenn du beobachtest, wie lange die Zelle braucht, um grünes Dendra2 wieder aufzufüllen und rotes Dendra2 loszuwerden, erfährst du, wie schnell diese Zelle neue Proteine (grün) herstellt und alte (rot) abbaut.
Bildnachweis: Life Science Databases
Natürlich ist uns Dendra2 eigentlich egal. Was wir wirklich wissen möchten, ist, wie Gehirnzellen mit dem normalen HD-Protein umgehen und ob dies durch die Mutation, die HD verursacht, verändert wird. Um dieser Frage nachzugehen, nutzten die Wissenschaftler Labortricks, um ein normales oder mutiertes HD-Protein mit dem Dendra2-Protein zu fusionieren. Nun konnten sie denselben Lichttrick anwenden, um dem HD-Protein zu folgen.
Der Müllmann kann
Mit diesem System stellten sie fest, dass mutiertes HD-Protein aus Neuronen im Striatum viel schneller beseitigt wurde als normales Htt. Das ist überraschend – viele Wissenschaftler hätten vorhergesagt, dass die mutierte Form des Proteins länger verbleiben würde und dass dieses Verweilen dazu führt, dass es sich zu Aggregaten ansammelt. Es deutet darauf hin, dass Neuronen in der Lage sind, das mutierte Protein zu erkennen und es zur Beseitigung zu markieren.
Tatsächlich ermöglichte das automatisierte Mikroskop, das Finkbeiners Team entwickelt hat, ihnen zu sehen, dass je schneller eine einzelne Zelle mutiertes Htt beseitigte, desto länger diese Zelle lebte. Das macht Sinn – das Aufräumen des toxischen Mülls schützt Gehirnzellen.
Wenn also das Erkennungs- und Eindämmungssystem für mutierte Proteine der Zelle funktioniert, wo liegt dann das Problem? Es stellt sich heraus, dass einige Neuronen besser in dieser Beseitigung sind als andere, nämlich die des Kortex. Der Kortex ist die faltige äußere Region des Gehirns. Im Allgemeinen erliegen Zellen im Kortex später der HD als Zellen im Striatum. In der Hoffnung zu verstehen, warum, verglichen die Autoren die Beseitigung des mutierten HD-Proteins im Striatum mit der Beseitigung im Kortex.
„Das Hauptergebnis dieser Studie ist, dass Neuronen aus verschiedenen Hirnregionen tatsächlich in ihrer Anfälligkeit für mutiertes Htt variieren. Es könnte eine Erklärung dafür liefern, warum HD an einer Stelle im Gehirn beginnt und nicht an einer anderen.“
Tatsächlich konnten Neuronen im Kortex mutiertes Htt schneller beseitigen. Neuronen des Kortex lebten auch länger als Neuronen des Striatums. Die Autoren schlussfolgerten, dass dies darauf zurückzuführen ist, dass diese Regionen Recyclingwege besitzen, die sich in ihren Fähigkeiten zur Beseitigung des mutierten HD-Proteins unterscheiden.
Wenn Proteine alt oder abgenutzt werden, verfügt die Zelle über ein System, um sie zu entsorgen oder zu recyceln. Die Forscher untersuchten, ob ihre Beobachtungen durch Auswirkungen auf dieses Abbausystem erklärt werden könnten. Insbesondere konzentrierten sie sich auf Nrf2, ein Protein, das den Recyclingweg einschaltet.
Wissenschaftler können untersuchen, wie verschiedene zelluläre Prozesse zu Dingen wie Zelltod und Recycling beitragen, indem sie das Volumen eines bestimmten Prozesses erhöhen oder reduzieren. In diesem Fall, weil sie glaubten, dass die durch das Nrf2-Protein aktivierten Recyclingprogramme wichtig sind, können sie den Zellen mehr Nrf2 geben oder es entziehen. Dies hilft ihnen festzustellen, ob ein spezifischer Prozess wichtig ist.
Genau das taten sie, um Nrf2 und das Recycling des mutierten HD-Proteins zu untersuchen. Neuronen, die mit zusätzlichem Nrf2 überladen waren, beseitigten mutiertes Htt schneller als normal und starben seltener ab. Dies deutet darauf hin, dass die Ankurbelung des zelleigenen Recyclingwegs ein potenzielles therapeutisches Ziel bei HD sein könnte.
Im Einklang mit dieser Idee waren die Zellen weniger in der Lage, mutiertes HD-Protein zu beseitigen, als die Wissenschaftler die primären Müll- oder Recyclingwege der Zelle künstlich mit Medikamenten reduzierten. Dieser spezifische Recyclingweg scheint also entscheidend zu sein, um Neuronen zu ermöglichen, mit diesem toxischen Protein umzugehen.

Bildnachweis: Chris Goodfellow
Was bedeutet das für die HK?
Das Hauptergebnis dieser Studie ist, dass Neuronen aus verschiedenen Hirnregionen tatsächlich in ihrer Anfälligkeit für mutiertes Htt variieren. Es könnte eine Erklärung dafür liefern, warum HD an einer Stelle im Gehirn beginnt und nicht an einer anderen. Der clevere Dreh ist, dass diese Anfälligkeit auf den unterschiedlichen Fähigkeiten dieser Neuronen beruht, die mutierten Proteine zu handhaben und zu entsorgen, anstatt auf einer intrinsischen Toxizität des Proteins selbst.
Dies ist tatsächlich sehr wichtig, da die Fähigkeit von Neuronen, mutiertes Htt zu handhaben, einen interessanten Weg für potenzielle Therapien eröffnet. Basierend auf den Ergebnissen dieser Studie würden wir vermuten, dass Behandlungen, die die Fähigkeit von Neuronen zur Beseitigung von mutiertem Htt steigern, Neuronen helfen würden, länger zu leben.
Diese interessante Möglichkeit muss jedoch im Kontext betrachtet werden. Erstens wurde diese gesamte Studie mit Neuronen durchgeführt, die in einer Schale gezüchtet wurden. Es bleibt abzuwarten, ob dieses Phänomen auch bei der menschlichen Krankheit beobachtet wird, was am wichtigsten ist.
Zweitens sind diese Ergebnisse vorläufig, da die oben beschriebenen möglichen Therapien noch nicht existieren. Es wird Zeit brauchen, etwas Passendes zu entwickeln und es in anderen HD-Modellen zu testen, bevor es am Menschen versucht werden könnte. Zum Beispiel weiß niemand, welche schädlichen Nebenwirkungen auftreten könnten, wenn der Nrf2-Weg über längere Zeit künstlich hochgefahren würde.
Unterm Strich sind diese Beobachtungen sehr faszinierende grundlegende HD-Wissenschaft. Und obwohl es noch keine zugelassenen Medikamente gibt, die das zugrunde liegende Problem bei HD angehen, tragen Studien wie diese zu einem besseren Verständnis dieser schrecklichen Erkrankung bei und ebnen den Weg für die Entwicklung neuer Medikamente.
Mehr erfahren
Weitere Informationen zu unseren Offenlegungsrichtlinien finden Sie in unseren FAQ…