
Huntington's Disease Therapeutics Conference 2021 – Tag 2
Unsere Übersicht zu Huntington's Disease Therapeutics Conference 2021 – Tag 2 #HDTC2021
Wir melden uns am 2. Tag der virtuellen CHDI Huntington’s Disease Therapeutics Conference 2021 zurück. Am Vormittag ging es diesmal um vielversprechende präklinische Forschung und am Nachmittag um unterschiedliche Biomarker der Huntington-Krankheit.
Welche neuen Medikamente gegen Huntington sind in der Pipeline?
Zusätzlich zu den laufenden klinischen Studien läuft auch die Grundlagenforschung zu Wirkstoffen gegen die Huntington-Krankheit weiter. Einige der interessantesten Projekte standen heute auf der Tagesordnung.
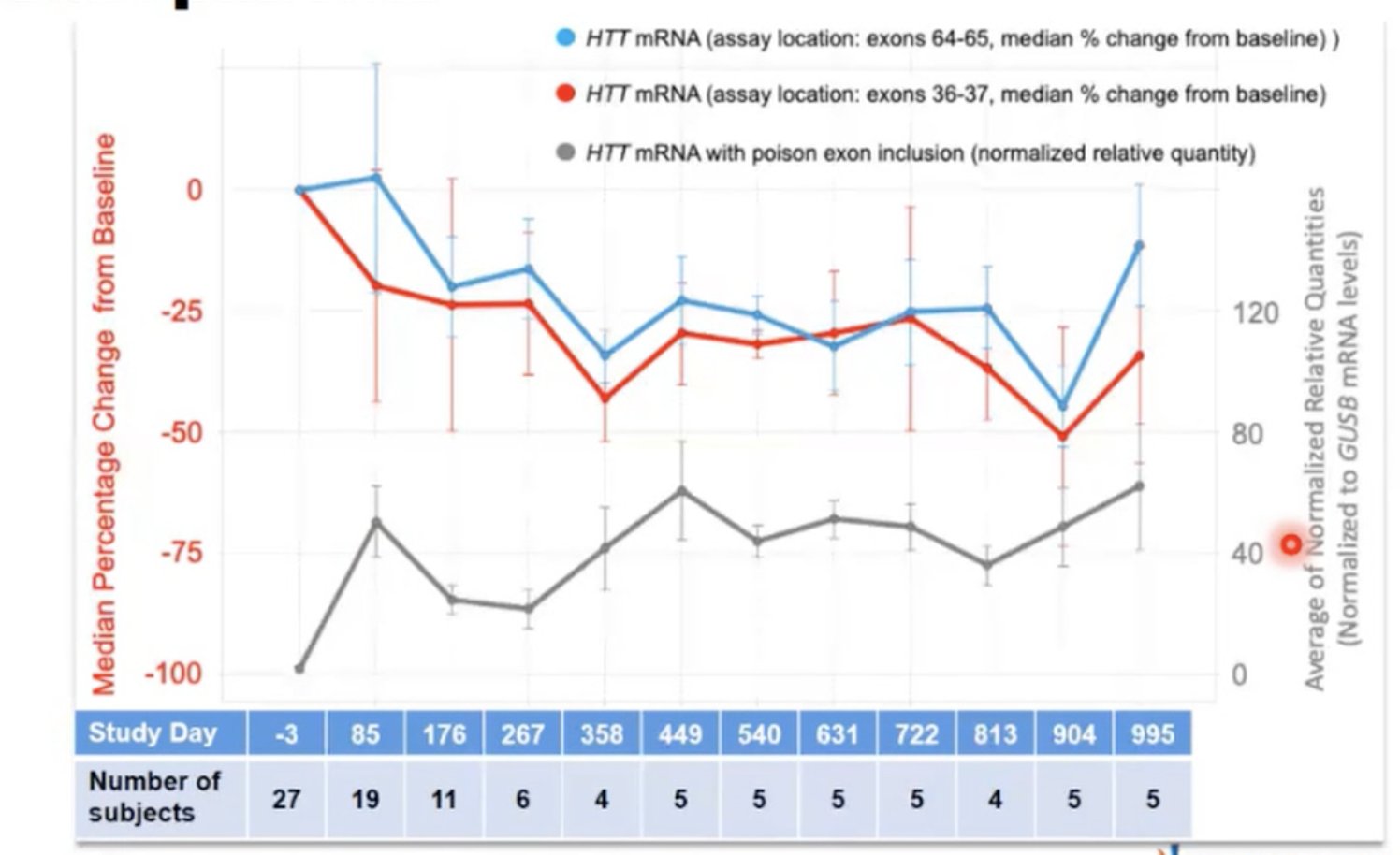
Image credit: Novartis
Reparatur des zellulären Entsorgungssystems: Entfernen giftiger Ansammlungen von Huntingtin
Der erste Vortrag kam von Peter Hamley von Samsara Therapeutics. Das Thema waren kleine Moleküle, die die Autophagozytose ankurbeln sollen, das ist eine Art von Müllentsorgung in der Zelle. Dieser Müll kann aus unnützen oder schädlichen Proteinhaufen, z. B. aus Huntingtin, bestehen. Das Ziel ist, die Zellen vor diesem Müll zu schützen, indem man mit Wirkstoffen die zelleigene Müllabfuhr unterstützt. Es ist bekannt, dass eine Verbesserung der Autophagozytose die Lebenserwartung von Tieren erhöhen kann, daher verfolgt man diesen Ansatz auch für die Behandlung altersbezogener bzw. degenerativer Erkrakungen wie der Huntington-Krankheit.
Samsara rastert hierbei systematisch Moleküle, um diejenigen zu finden, die Autophagozytose begünstigen und schaut sich im Folgenden genauer an, ob sie auch dazu führen, dass Proteinklumpen besser entfernt werden. Mithilfe ihrer Moleküldatenbank konnten Samsara bereits einige interessante Verbindungen identifizieren, die sich in Zellkulturen und in Mäusen günstig auf die Autophagozytose auswirken. Diese scheinen besser als bisher bekannte Stoffe zu wirken und zeigen sich bereits vielversprechend bei anderen Formen der Neurodegeneration, z. B. bei Alzheimer. Die Anwendung dieser Erkenntnisse auf die Huntington-Krankheit geht Samsara schrittweise an und untersuchte zunächst Huntington-Zellkulturen und -Mücken. Sie rechnen damit in Kürze den besten Kandidaten definieren zu können und hoffen auf dessen weitere Testung als Huntington-Medikation im Jahr 2022.
Kleine Moleküle zur Huntingtin-Verminderung
Als Nächste trug Beth Hoffman von Origami Therapeutics vor. Passend zum Namen beschäftigt sich das Unternehmen mit dem richtigen und schädlichen Falten von Eiweißen. Durch die extra-lange CAG-Sequenz faltet sich das mutierte Huntingtin zu einer anderen Form zusammen als der Wildtyp. Dies könnte ein Grund sein, warum es daraufhin in schädliche Eiweißbestandteile zerlegt wird. Origami Therapeutics entwickelt kleine Moleküle, die sich um die Entsorung dieser Bestandteile kümmern sollen. Die Moleküle könnten entweder die Entsorgungsmechanismen der Zelle stimulieren oder auch die Struktur des gefalteten Eiweißes stabilisieren, sodass keine ungesunden Abbauprodukte entstehen.
Origami misst hierfür die Anhäufung des mutierten Eiweißes und die Schädigungen, von denen sie annehmen, dass sie durch die Verklumpungen verursacht werden.
Austausch erkrankter Hilfszellen im Gehirn
Steve Goldman, ein Forscher, der sich mit Heilungsstrategien durch Ersatz erkrankter Gliazellen mit gesunden Gliazellen im Gehirn beschäftigt, hielt den nächsten Vortrag.
Im Rahmen seiner Forschung will er herausfinden, ob der Verlust an Gliazellen durch die Huntington-Mutation auch Auslöser für Symptome der Krankheit ist. Durch Einpflanzen von Gliazellen mit der Huntington-Mutation in Mäusegehirnen, konnte seine Gruppe zeigen, dass die Mäuse Symptome und abnormales Verhalten entwickelten. Auch in Zellkulturen in der Petrischale lässt sich beobachten, dass bei Gliazellen mit der Mutation ganz andere Gene aktiv bzw. inaktiv sind als bei Gliazellen ohne die Mutation. Gehirnzellen, die von Huntington-Gliazellen umgeben sind zeigen eine schlechtere Funktionalität. Astrozyten sind eine bestimmte Art von Gliazellen, die bei Anwesenheit der Mutation Veränderungen in ihrer Form, Entwicklung und Gesundheit zeigen.
Goldman’s Gruppe wollte herausfinden, ob die Transplantation von gesunden Gliazellen in Huntington-Mäuse zu einer Verbesserung ihrer Symptome führen kann. Sie konnten tatsächlich abgemildert Symptome und eine höhere Lebenserwartung der Mäuse beobachten. Goldmann hat sich zum Ziel gesetzt, solche Transplantationen auch bei Menschen durchzuführen und bereitet derzeit entsprechende klinische Studien vor.
Von spinaler Muskelatrophie zur Huntington-Krankheit
„Zusätzlich zu den laufenden klinischen Studien läuft auch die Grundlagenforschung zu Wirkstoffen gegen die Huntington-Krankheit weiter.“
Der Themenabschnitt wurde durch den Vortrag von Rajeev Sivasankaran von Novartis abgerundet. Er handelte von Branaplam, ein Huntingtin-Verminderungs-Medikament, dass man als Tablette schlucken kann. Wir berichteten in einem unserer letzten Artikel schon darüber.
Novartis hatte bisher an einer anderen neurodegenerativen Erkrankung geforscht, der spinalen Muskelatrophie (SMA), die ausgeprägte Muskelschwäche bei Kindern auslöst. Auch SMA ist eine Erbrankheit, allerdings wird sie durch ein fehlendes Gen/Protein hervorgerufen und nicht wie die Huntington-Krankheit durch ein vorhandenes mutiertes Gen. Das Medikament Branaplam von Novartis stellt das fehlende Eiweiß her, indem es einen Trick namens „Splice Modulation“ (~Verbindungsmodulation) anwendet.
Durch Branaplam wird ein Gen in den Zellen aktiviert, dass ein Eiweiß erzeugt, das die Funktion des fehlenden Eiweißes ersetzen kann. Bei SMA-Kindern konnten erste klinische Studien schon Erfolge vorweisen.
Genauer gesagt, wirkt Branaplam auf die Verbindungen, die durch die Boten-RNA hergestellt werden. Auf diese Weise könnte es auch die Herstellung des Huntingtins beeinflussen. Durch einen glücklichen Zufall weist das Huntington-Gen nämlich die gleiche Sequenz auf und mit deren Hilfe könnte Branaplam die Herstellung des Proteins drosseln. Es führt dazu, dass ein Abschnitt in die mRNA eingefügt wird, der normalerweise abgebaut werden würde. Diese mRNA weist dann die Zelle nicht mehr dazu an, mehr Eiweiß zu produzieren sondern sagt ihr das Gegenteil.
Bei Huntington-Mäusen konnte die Wirsamkeit von Branaplam bereits gezeigt werden. Sivasankaran zeigte die Daten dazu. Drei Wochen lang nach der Gabe von Branaplam konnte eine geringere Konzentration von Huntingtin bei ihnen gemessen werden, danach stieg das Niveau von Huntingtin wieder an.
Anhand der Daten aus der klinischen Studie mit den SMA-Kindern konnten ebenfalls Absenkungen von Huntingtin beobachtet werden. Huntingtin wurde in etwa um ein Drittel vermindert und die Konzentration blieb niedrig. Novartis ist bereits weit fortgeschritten mit der Planung einer klinischen Studie zu Branaplam und der Huntington-Krankheit, auch wenn heute keine Details dazu bekannt gegeben wurden. In der anschließenden Fragerunde sprach Sivasankaram jedoch davon, dass weitere Informationen dazu in den nächsten Wochen und in diesem Sommer bekannt gegeben werden sollen.
Biomarker zur Überprüfung der Huntingtin-Verminderung
Am Nachmittag lag der Fokus auf Biomarkern. Mithilfe von Biomarkern sind Wissenschaftler und Ärzte in der Lage, auf der einen Seite das Fortschreiten der Huntington-Krankheit und auf der anderen Seite die Wirksamkeit von Medikamenten zu überprüfen.
Bildgebende Verfahren zur Erfassung des Huntingtins im Gehirn
Daniele Bertoglio berichtete von seiner Forschung an der Erfassung von Huntingtin im Gehirn durch bildgebende Verfahren. Seine Gruppe arbeitet an ausgeklügelten Werkzeugen um dieses Ziel zu erreichen.
Derzeit kann die Menge an Huntingtin im Gehirn nur indirekt bestimmt werden, indem man Nervenwasserproben am Rücken entnimmt. Näheres dazu finden Sie bei Interesse hier. Um eine direkte und gleichzeitig nicht-invasive Messung im Gehirn zu ermöglichen, arbeitet Bertoglios Gruppe gerade an Verfahren zur chemischen Markierung des Huntingtins. Diese chemischen Stoffe werden auch Tracer genannt und enthalten schwach radioaktive Elemente, die über Positionen-Emissions-Tomographie (PET) erfasst werden können. PET ist eine häufig eingesetzte, medizinische Methode, bisher fehlen lediglich die für Huntingtin passenden Tracer.

An Huntington-Mäusen, die mit Huntingtin-Verminderungsmedikamenten behandelt wurden, hat Bertoglio’s Gruppe ihren eigens entwickelten Tracer getestet. Die PET zeigte schwächere Signale bei Mäusen mit gerinegeren Huntingtin-Werten, so wie es sich die Forscher erhofft hatten. Es konnten sowohl niedrigere Huntingtin-Gesamtwerte als auch lokal niedrigere Huntingtin-Konzentrationen mit unterschiedlichen Techniken der PET erfolgreich sichtbar gemacht werden.
In der anschließenden Fragerunde erwähnte Bertoglio noch, dass seine Gruppe den neuen Tracer auch an gespendetem Hirngewebe von Huntington-Patienten testen konnte, wo sich die Funktionalität zu bestätigen schien.
Erprobung von Huntingtin-Tracern im Menschen
Der im vorangegangenen Vortrag beschriebene Tracer ist tatsächlich schon Kern einer laufenden klinischen Studie namens iMagemHTT, von der uns Andrew Wood in seinem Vortrag berichtete. Hier werden zu gleichen Zeitpunkten Blutproben und PET/MRT-Scans untersucht, um die Funktionalität des Tracers und seine Wechselwirkungen im Körper zu prüfen.
Wood untersuchte zunächst verschiedene Dosierungen des Tracers und dessen Verweildauer im menschlichen Körper. Mit der scheinbar geeignetsten und sichersten Dosis wurde im Anschluss fortgefahren. Er stellte vorläufige Daten von drei Teilnehmern einer Phase-I-Studie vor, es wurden die Verträglichkeit und die Abbaugeschwindigkeit untersucht, bisher gab sieht es für den Tracer gut aus.
Die Studie läuft noch im nächsten Jahr weiter, es werden Menschen mit und ohne die Huntington-Krankheit freiwillig teilnehmen, sie erhalten den Tracer und es werden Blutproben abgenommen und PET/MRT-Scans durchgeführt. Im Hintergrund arbeitet Wood’s Gruppe zusammen mit der CHDI an weiteren Tracer-Kandidaten und untersucht deren Eigenschaften, um im Zweifelsfall darauf zurückgreifen zu können.
Da die Huntingtin-Konzentration im Nervenwasser unter Umständen nicht der Huntingtin-Konzentration im Gehirn entspricht, liegen hohe Erwartungen auf diesen Tracern, um zukünftig eine sichere Diagnose machen zu können. Wood hofft, dass es einer der Kandidaten im Laufe des Jahres 2022 in die Kliniken schaffen könnte.
Untersuchung von Huntington-Biomarkern an nicht-menschlichen Huntington-Primaten
Jodi McBride sprach als Nächste von ihrer Forschung zu Biomarkern an nicht-menschlichen Primaten mit mutiertem Huntington-Gen. Viele Huntington-bezogenen Studien arbeiten mit gesunden Affen, um beispielsweise nachvollziehen zu können, wie Wirkstoffe sich in einem großen Gehirn verteilen können. McBride hat eine neue Methode entwickelt, bei der sie mithilfe eines Virus das Huntington-Gen in die Gehirne von Affen einbringt. Das Gen sorgt für die Herstellung von Huntingtin, es bilden sich Eiweißablagerungen. Die Affen weisen Probleme im Arbeitsgedächtnis und bei Bewegungen sowie Veränderungen im Gehirn auf, ähnlich wie sie von der Huntington-Krankheit bei Menschen bekannt sind.
McBride’s Gruppe ist weiterhin damit beschäftigt, bildgebende Verfahren und andere Methoden an den Affen zu testen, um festzustellen, ob sie sich für die Erforschung von Biomarkern für die Huntington-Krankheit als Tiermodell eignen.
Innovationen in der Bildgebung, die das Fortschreiten der Huntington-Krankheit sowie neue Biomarker sichtbar machen
„Mithilfe von Biomarkern sind Wissenschaftler und Ärzte in der Lage, auf der einen Seite das Fortschreiten der Huntington-Krankheit und auf der anderen Seite die Wirksamkeit von Medikamenten zu überprüfen.“
Derek Jones war nun an der Reihe, um über neue MRT-Techniken zu sprechen. Er betont sowohl die Möglichkeiten als auch die Herausforderungen dieser nicht-invasiven Art der Untersuchung.
Viele unterschiedliche Veränderungen im Gehirn können für den Laien gleich aussehen, sie auseinanderzuhalten ist für die richtige Interpretation essentiell. Die technologische Verbesserung der MRT-Geräte ermöglicht ein leichteres Erkennen der Unterschiede und solche neuen Geräte werden in den Krankenhäusern immer gängiger. Durch sie werden auch Bereiche im Gehirn sichtbar, die vorher kaum erfasst werden konnten, die aber besonders wichtig für den Verlauf der Huntington-Krankheit sind, wie z. B. das Striatum.
In Jones‘ Labor werden Methoden für Untersuchungen entwickelt, die es den behandelnden Ärzten erlauben, so viele Informationen wie möglich aus den MRT-Scans zu erhalten. Dahinter steckt komplizierte Mathematik. Es können unter anderem sehr subtile Veränderungen bei Huntington-Betroffenen, die noch keine Symptome zeigen, sichtbar gemacht werden.
Jones bedankte sich für die Arbeit im Rahmen der HDClarity-Studie, die eine Standardisierung für die Entnahme von Nervenwasserproben von Huntington-Patienten entwickelt hat. In einem Project namens ImageClarity wendet er die gleichen Prinzipien zur Standardisierung für seine Scans an.
Neue Biomarker im Nervenwasser
Niels Skotte hielt heute die letzte Präsentation. Seine Forschung dreht sich um die Quantifizierung verschiedener Eiweiße im Nervenwasser und im Blut von Huntington-Patienten. Er hält Ausschau nach neuen Biomarkern, die das Fortschreiten der Krankheit sichtbar machen sollen.
In winzigen Mengen Nervenwasser oder Blut können seine Kollegen derartige Eiweiße identifizieren. Sie beobachten in beiden Medien signifikante Veränderungen, wenn sie gesunde Kontrollproben mit denen von Huntington-Patienten mit und ohne Symptome vergleichen.
Ein beispiel ist das Protein NfL, das schon früher wissenschaftlich beschrieben wurde. Die Konzentration von NfL nimmt zu, wenn die Krankheit weiter voranschreitet. Daher wird es bereits in klinischen Studien als Biomarker verwendet. Ein weiteres Protein, mit dem sich Skotte beschäftigt trägt den Kurznamen PENK. Dessen Menge verringert sich mit fortschreitender Erkrankung. Hier forscht seine Gruppe weiterhin an der genauen Korrelation mit der Krankheit.
Bis morgen!
Im nächsten Artikel werden wir vom dritten Tag der Konferenz berichten.
Weitere Informationen zu unseren Offenlegungsrichtlinien finden Sie in unseren FAQ…


