Die Vorteile der Migration, hervorgehoben bei der Huntington-Krankheit
Ist HD eine Entwicklungsstörung? HD stoppt die Migration von Neuronen im sich entwickelnden Gehirn, aber vielleicht können wir sie wieder in Gang bringen
Huntingtin, das für die Huntington-Krankheit verantwortliche Protein, ist für die Entwicklung von Föten im Mutterleib von grundlegender Bedeutung, aber wir wissen noch nicht genau, welche Rolle es in diesem komplexen Prozess spielt. Normalerweise beginnen Neuronen ihr Leben tief im sich entwickelnden Gehirn, wandern an die Oberfläche und bilden dann ein Netzwerk von Verbindungen mit anderen, aber Sandrine Humberts Gruppe zeigte, dass diejenigen ohne Huntingtin stecken bleiben und nie dorthin gelangen, wo sie hingehören. Neuronen mit mutiertem Huntingtin sind nicht besser als solche, denen es vollständig fehlt. Die Wiedereinführung von normalem Huntingtin oder der Proteine, über die es wirkt, ermöglicht es Neuronen jedoch, wieder normal zu migrieren, was verlockende neue Wege zur Behandlung der Huntington-Krankheit eröffnet.
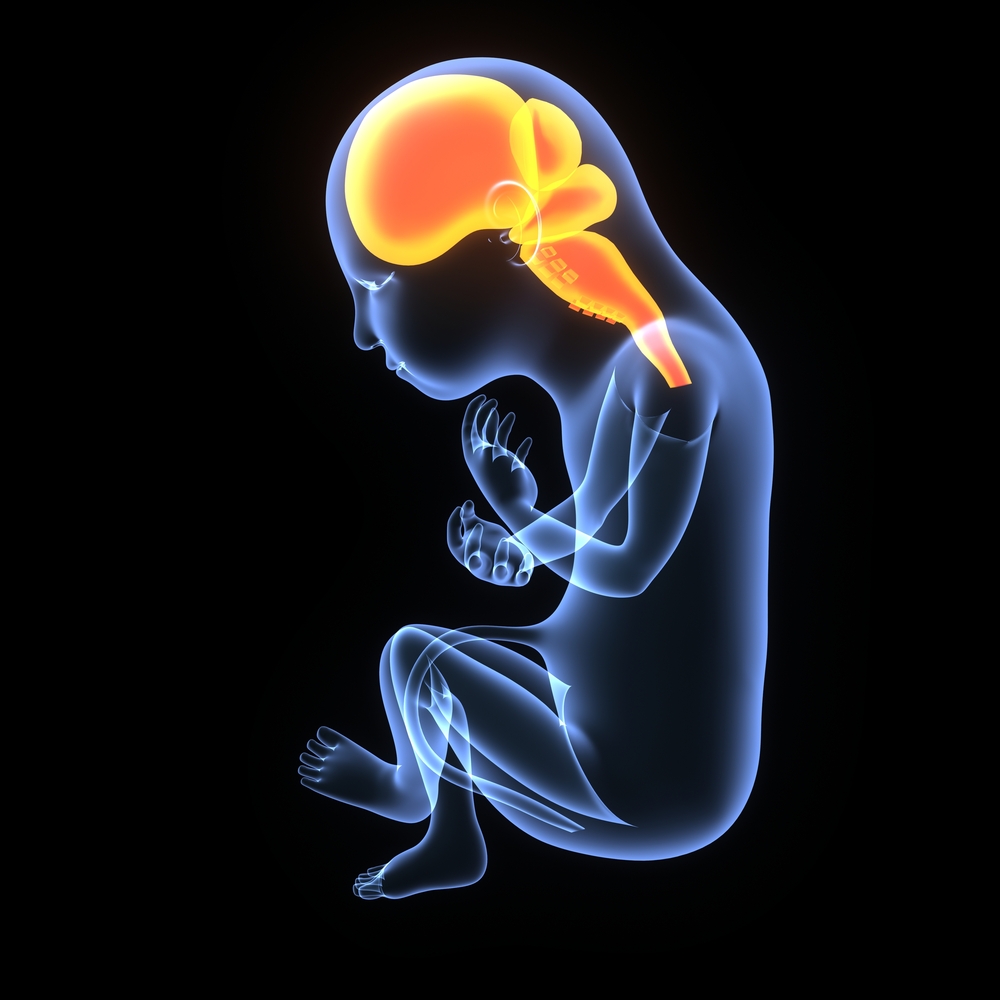
Wie entsteht ein Gehirn?
Wir alle beginnen unser Leben als einzelne Zelle, wenn die Eizelle unserer Mutter von der Samenzelle unseres Vaters befruchtet wird. Diese Zelle teilt sich immer wieder, wird zuerst zu einem runden Zellklumpen und entwickelt sich dann zu einer wurmartigen Struktur, die Embryo genannt wird. Entlang des Rückens des Embryos faltet sich ein schmaler Streifen nach innen und bildet einen Schlauch, der von oben nach unten verläuft. Dieses „Neuralrohr“ bildet unser Nervensystem – unser Gehirn, Rückenmark und all unsere Nerven. Die Wand des Schlauchs hat mehrere Schichten. Es ist die innerste, nahe dem flüssigkeitsgefüllten Zentrum, wo Nervenzellen, Neuronen genannt, entstehen.
Jedes Neuron streckt zwei fingerartige Fortsätze aus, einer zur äußeren Oberfläche des sich entwickelnden Gehirns und einer, der zum Zentrum des Schlauchs zeigt. Durch diese Veränderung ausgelöst, bewegen sich Neuronen zur äußeren Oberfläche und reifen dabei. Diesen Vorgang nennen wir Migration. Schließlich wird die äußere Schicht des Gehirns mit Neuronen gefüllt. Diese Schicht wird Kortex genannt. Sobald Neuronen die Gehirnoberfläche erreichen, bilden sie weitere kleine Fortsätze, die Kontakt zu anderen Neuronen aufnehmen, um Signale zu kommunizieren.
Der Kortex ist entscheidend für all unsere Denkprozesse, wobei verschiedene Teile unterschiedliche Aufgaben wie Empfindung, Bewegung und Persönlichkeit übernehmen. Krankheiten, die die Gehirnentwicklung stören, werden als neurodevelopmentale Störungen bezeichnet, und sie können zu Veränderungen in der Gehirnstruktur führen, die Denkprozesse beeinträchtigen oder Anfälle verursachen.
Wie beeinflusst das Huntingtin-Protein die Gehirnentwicklung?
Wir wissen bereits, dass das Huntingtin-Protein für die Bildung eines Embryos wichtig ist, da Mausembryonen mit niedrigen Huntingtin-Spiegeln Defekte in ihren Nervensystemen aufweisen und diejenigen, denen es vollständig fehlt, nicht einmal bis zur Geburt überleben. Wir wissen jedoch nicht, was Huntingtin im sich entwickelnden Embryo tatsächlich tut, das so wichtig ist! Sandrine Humberts Gruppe in Frankreich hat genau das untersucht.
In Mausembryonen, sehr früh in der Entwicklung, schaltete ihre Gruppe das Huntingtin-Gen in Neuronen aus. Obwohl die Neuronen normal reiften, entwickelten sie diese beiden Fortsätze nicht und migrierten nicht zur Gehirnoberfläche, sodass der Kortex dünner wurde. Viele Neuronen blieben in den tieferen Schichten des Gehirns stecken und erreichten nie den Kortex. Selbst diejenigen, die den Kortex erreichten, sahen nicht normal aus und bildeten weniger Verbindungen zu anderen Neuronen. Diese Defekte besserten sich mit der Zeit nicht und waren immer noch vorhanden, als die Mäuse erwachsen wurden.
Die Inaktivierung von Huntingtin in einem späteren Stadium, nachdem Neuronen migriert waren, beeinflusste die Dicke des Kortex nicht, begrenzte aber immer noch die Verbindungen, die die Neuronen herstellten.
„Wir haben jetzt eine bessere Vorstellung davon, warum Huntingtin im sich entwickelnden Embryo so wichtig ist, und dieses Wissen könnte uns in Zukunft zu neuen Behandlungen für die Huntington-Krankheit führen.“
Die Gruppe führte dann das normale Gen wieder in diese Neuronen ein und zeigte, dass sie dann wieder normal migrieren konnten.
Wir haben also jetzt noch mehr Beweise dafür gesehen, wie wichtig Huntingtin für die Gehirnentwicklung ist, aber wir wissen immer noch nicht genau, wie es das tut.
Wie genau steuert Huntingtin die Gehirnentwicklung?
Es ist bekannt, dass Huntingtin mit einem anderen Protein namens RAB11 interagiert, das steuert, wie sich Substanzen im Neuron bewegen. Ein solches Molekül, das zu den wachsenden Armen migrierender Neuronen transportiert wird, ist N-Cadherin, das bekanntermaßen wichtig für die Entwicklung des Nervensystems ist.
Als Humberts Gruppe Huntingtin inaktivierte, blieb N-Cadherin im Zentrum der sich entwickelnden Neuronen stecken und wurde nicht an seinen normalen Ort an der Vorderkante der migrierenden Zelle transportiert. Als den Neuronen jedoch gesagt wurde, RAB11 zu produzieren, gelangte N-Cadherin zur Vorderkante. Das bedeutet, dass wir einige der molekularen „Gehilfen“ identifiziert haben, die Huntingtin verwendet, und indem wir diese ersetzen, können wir die normale Gehirnentwicklung wiederherstellen.
Humberts Gruppe hat also begonnen, die normale Funktion von Huntingtin im sich entwickelnden Gehirn zu entschlüsseln. Menschen mit Huntington-Krankheit fehlt jedoch kein Huntingtin. Sie produzieren das Protein immer noch, aber es ist eine Version, die Neuronen schädigt. Wie ist das also für HD relevant?

Was ist mit mutiertem Huntingtin?
Wie wir bereits hörten, stoppt das Ausschalten von Huntingtin in Mausembryonen die Migration von Neuronen zur Gehirnoberfläche. Wie erwartet, ermöglicht die Wiedereinführung von normalem Huntingtin den Neuronen, ihr Ziel zu erreichen. Als sie jedoch stattdessen mutiertes Huntingtin einführten, blieben die Neuronen in den tiefen Schichten stecken. Das deutet darauf hin, dass das mutierte HTT-Protein einen Teil seiner normalen Funktion in der Gehirnentwicklung verloren hat.
Verursacht also eine abnormale Gehirnentwicklung Symptome der Huntington-Krankheit?
Humberts Gruppe fand heraus, dass die Inaktivierung von Huntingtin in sich entwickelnden Neuronen verhindert, dass diese die richtige Form annehmen, den korrekten Ort im Gehirn erreichen und Verbindungen mit anderen Neuronen bilden. Mutiertes Huntingtin hatte einen ähnlichen Effekt. Dies zeigt, dass Huntingtin eine Schlüsselrolle in der Gehirnentwicklung spielt, aber es tut dies nicht allein … es wirkt, indem es den Transport wichtiger Proteine zur Vorderkante migrierender Neuronen steuert. Wichtig ist, dass man, wenn man diese Proteine ersetzen kann, die normale neuronale Entwicklung wiederherstellen kann.
Wir haben die Huntington-Krankheit traditionell als eine Erkrankung mit Beginn im Erwachsenenalter betrachtet, da die Symptome normalerweise dann beginnen. Sollten wir sie jedoch angesichts dieser neuen Erkenntnisse stattdessen als neurodevelopmentale Erkrankung betrachten? Wir wissen sicherlich, dass Scans subtile Veränderungen in den Gehirnen von Mutationsträgern ein Jahrzehnt oder länger vor dem Auftreten der Symptome aufzeigen können. Andererseits gibt es nicht viele Beweise dafür, dass menschliche Gehirne vor dem HD-Beginn die hier beschriebenen Probleme bei der Neuronenmigration zeigen. Um schnell Antworten zu erhalten, werden Mausmodellen tendenziell extreme Veränderungen gegeben, die bei Menschen nie beobachtet werden – totale Proteindeletionen oder riesige HD-Mutationen. Wenn etwas Ähnliches in den sich entwickelnden Gehirnen von Menschen mit einer HD-Mutation vor sich geht, ist es wahrscheinlich viel subtiler – aber diese Arbeit kann uns helfen, es zu finden und zu untersuchen, und es vielleicht nutzen, um neue Medikamente zum Schutz vor mutiertem Huntingtin zu entwickeln.
Wir haben also jetzt eine bessere Vorstellung davon, warum Huntingtin im sich entwickelnden Embryo so wichtig ist, und dieses Wissen könnte uns in Zukunft zu neuen Behandlungen für die Huntington-Krankheit führen. Es liefert uns auch wichtige Informationen, die uns helfen werden zu entscheiden, wie und wann wir die Verabreichung von „Gen-Silencing“-Medikamenten in Betracht ziehen sollten, um sicherzustellen, dass der Nutzen des Ausschaltens von mutiertem Huntingtin jedes Risiko durch die Senkung des „normalen“ Proteins überwiegt.
Mehr erfahren
Weitere Informationen zu unseren Offenlegungsrichtlinien finden Sie in unseren FAQ…